2020-02-06 12:46:08
I. V. Kochikov1 & A. V. Stepanova & G. M. Kuramshina
Introduction
Quantum mechanical calculations of indole and pyrrole molecules w ere p erformed with the p rogram GAUSSIAN 03 (Revision B.03) package. In the practice of processing experimental data obtained by means of infrared and Raman spectroscopy, a number of var- ious mathematical inverse problems arise. The most important of them is the so-called inverse vibrational problem where parameters of the molecular force field (force constants) are determined from given experimental data (vibrational fre- quencies, isotope frequency shifts, Coriolis constants, centrif- ugal distortion constants, etc.). Force constants are indispens- able for prediction of spectra and other properties of compounds not yet investigated and for development of phys- ical models in a theory of molecular structure.
The general inverse vibrational problem is formulated as nonlinear operator equation in finite-dimensional spaces
AF ¼ Λ where A is an operator completely defined by available mo- lecular configuration, vector Λ∈Rm represents the set of avail- able experimental data (vibrational frequencies, etc.) and F is an unknown symmetrical force constant matrix, F∈ Rn(n + 1)/2. Experimental data as well as operator A are known with certain errors, that is, instead of A and Λ, we are given Ah and Λδ where ||Λ–Λδ|| ≤ δ and h is a numerical estimate of the operator A uncertainty: Ah F ¼ Λδ
The problem of calculating molecular force fields from the frequencies of normal vibrations of a molecule is an ill-posed problem; it does not satisfy all three well-posedness condi- tions (existence of solution, its uniqueness, and stability to perturbations in input data) [1]. Indeed, for any molecular structure (except diatomic molecules), there may exist an infinite number of force field matrices which result in the same normal vibration frequencies. In the most general formulation, the stable solution of problem (1) may be obtained on the base of Tikhonov regularization method [2–4] that allows to utilize additional information on the solution properties. The main idea of this approach is that solution should be in some respect close to a priori given force constant matrix F0. This a priori given matrix should be either based on some model assump- tions, e.g., synthesized form the known force constants of related molecules or, preferably, obtained from quantum- chemical calculations [5–8]. In the framework of this ap- proach, the stable solution may be obtained as an extremum Fα of Tikhonov’s functional on the set of constraints D where F0 is some a priori given stabilizing quantum-mechanical matrix. In frame of Tikhonov’s regularization technique, the regularization pa- rameter α should be chosen with account to the input data errors: α = α (h, δ) [3, 5]. As a result, we obtain matrix Fα(h,δ) that is closest in the sense of Euclidian norm to a given matrix F0 among the set of solutions of Eq. (1) compatible with experimental data within the error level δ. The optimized solution is referred to as regularized quantum mechanical force field (RQMFF) [5–8]. The regularizing procedure allows using any system of generalized coordinates, including redun- dant system of internal coordinates, and various constraints on the values of force constants which allow to narrow the set of possible solutions. Since definition of the force-field matrix in a redundant coordinate system is not unique, two conventions were introduced (1) that the off-diagonal norm of matrix of force constants in redundant coordinates be a minimum; this choice is well in accord with the commonly used force field models [5] and (2) the choice of the so-called canonical matrix [5]. The same statement of inverse problem was realized for the case of special parametrization of the force constant matrix via scaling factors [6].
In 1998, Professor Victor Spiridonov has initiated collabo- ration between two laboratories of the Department of Physical Chemistry (molecular spectroscopy and electron diffraction) and the RQMFF concept was applied to the determination of equilibrium geometry and harmonic force field of free mole- cules by the joint use of electron diffraction, vibrational spec- troscopy, and ab initio data for the benzene molecule [9]. As a result, a practical program package for the integrated analysis was developed. In 1999, this integrated algorithm was extended to include systems with large-amplitude motion [10, 11]. Additionally, the treatment was augmented by the inclusion of microwave data (rotational constants) and centrifugal distortion corrections to interatomic distances caused by the rotational motion of a molecule to eliminate the source of trouble in the analysis of diffraction data collected at elevated temperatures.
In [10] the force field constants were expressed in the internal coordinates and fitted via scale factors obtained by the regular- ized least-squares refinement to ensure the best fit between calculated and observed spectroscopic or diffraction evidence. The developed algorithms are widely used in the investigations of molecular structure and vibrational spectra of polyatomic molecules, some examples are presented in Refs. [11–14].
In the practice of spectroscopic data analysis, the first at- tempts to find a compromise between the cost of ab initio calculations and the desire of investigators to have good agreement between ab initio and observed vibrational fre- quencies were initiated in Ref. [15]. The further progress in this direction was connected with appearance of very effective approach for obtaining force constants compatible with exper- imental data; the so-called Pulay scale factors method [16] in which discrepancy ‖AhF − Λδ‖ is minimized on the set of force constant matrices D such that D ¼ F : F ¼ BF0B ; B ¼ diagfβ1; …; βng ð3Þ where B is a diagonal matrix of scale factors, and F0 is a priori given matrix. Though constraints Eq. (3) do not necessarily allow obtaining solution that is compatible with experimental data within experimental errors, the scaling procedure has become an important instrument for the practicing spectrosco- pists, extremely popular due to its simplicity. Besides, it has been shown that for many molecular fragments, the scale fac- tors (within a given level of quantum-mechanical method) are approximately constant in a wide range of similar molecules. To date, the force constant scaling factors for different levels of theory have been obtained, which in most cases allow to approximate experimental frequencies with a reasonable de- gree of accuracy.
Note that parameterization Eq. (3) does not completely remove the ambiguity of the solution of Eq. (1), but it may be resolved by using the same regularization scheme on the set D by searching for the matrix F closest to F0, or by searching for the scale matrix B closest to unit matrix [14]. Solution in terms of scale factors is obtained as an extremum of Tikhonov’s functional Eq. (2) [6, 14].
Nowadays, the scaling in internal coordinates is very popular as very convenient and easy way of fitting calculated frequencies to observed ones. The scale factors method has been originally suggested and implemented for the force fields defined in the internal or symmetry (local symmetry) coordi- nates. The useful recommendations on the choice of internal coordinates for different molecules are given in Ref. [17]. Some numerical aspects of determination of scaling factors in frame of theory of regularization of ill-posed problem were considered in Ref. [18]. Nevertheless, in practice, for the huge molecules/molecular systems introduction of the complete system of internal coordinates is often a tedious, time-consum- ing, and not uniquely defined procedure.
In a case of rather large systems, there can be many specific situations where Cartesian space can actually be more efficient than using other generalized coordinates and the scaling factors expressed in terms of internal co- ordinates are the certain stipulation for the correction of results. In such cases, the choice of the Cartesian coordi- nates for constructing a force field from fragments re- mains much simpler procedure than using the internal coordinates [19, 20].
Quantum-chemical calculations usually provide the force constant matrix in Cartesian coordinates. It is tempt- ing to formulate a scaling procedure that would avoid introducing any alternative generalized coordinates. On this way, we have proposed the procedure [20] for scaling a quantum-chemical force–field matrix in Cartesian coor- dinates within a general approach of scaling that implies calculation of scale factors for the moderate-size molecules when quantum-chemical calculations are possible.
Cartesian coordinates may be then used in investigating large molecules when accurate quantum-chemistry calcu- lations are impossible or extremely time-consuming. The results of scaling can be used in the following ways:
1. For predicting IR and Raman spectra of the large molecules or clusters when quantum-chemistry calculations are inaccessible. In this case, it is possible to obtain the quick estimates of vibrational spectra by the synthesis of the force constant matrix from separate blocks of the scaled force constants.
2. For improvement of the calculated spectra of the large molecular systems that allow only a low-level quantum chemical calculation (e.g., DFT methods). In this case, scale factors obtained for the same method for the smaller molecules may be applied to correct the calculated force matrix.
These capabilities can be realized through the scaling both in terms of internal coordinates and in terms of Cartesian coordinates. In this respect, the suggested ap- proach is not significantly different from the conventional scaling procedures. Both of them are aimed at investigat- ing huge molecules (clusters, complexes, etc.); we have many examples when the numerical calculation of fre- quencies required several months of computing.
Alongside with that, the problem of scaling in the Cartesian coordinates, however, is nontrivial, for exam- ple, the scale matrix B determined for Cartesian coordi- nates can no more be considered diagonal. Nevertheless, it proved possible to formulate certain conditions allowing to find appropriate scale factors using Cartesian coordinates [20]. In the next section, we brief- ly recall the main results.
Scaling ab initio molecular force fields in Cartesian coordinates
The main problem in scaling force-field matrix in the Cartesian coordinates is that potential energy of a mole- cule defined by this matrix is not automatically indepen- dent of the molecular position and orientation as in the case of internal coordinates. As a result, a physically meaningful force constant matrix should satisfy certain conditions to eliminate translational and vibrational de- grees of freedom in the expression of the potential energy.
Constraints Eq. (5) reduce the rank of matrix F to 3 N-6 (or 3 N-5 for linear molecules), thus leaving only vibrational de- grees of freedom. It is easy to see that matrix F obtained from F0 by scaling procedure Eq. (3) with a diagonal matrix B does not necessarily satisfy constraints Eq. (5), even if the orig- inal matrix F0 does satisfy them. To ensure that the scaled matrix F also describes only vibrational degrees of free- dom, the scale matrix B should satisfy certain conditions. Following [20], we assume that scaling in Cartesian coor- dinates is still performed by Eq. (3), but the matrix B is no more diagonal and should have the following properties:
1. Matrix B consists of the 3 × 3 unit submatrices multiplied by certain factors βij (i, j, = 1, …, N):
2. The factors βij satisfy the following conditions:
It is easy to see that conditions Eq. (7) allow matrix B to be diagonal only when all βii are equal.
The described algorithm was successfully applied to vari-where Fm ¼ Bm F0 Bm. The resulting matrix is calculated ous molecules.
Solution of the direct and inverse vibrational problems is often facilitated by the presence of certain molecular sym- metry. Due to symmetry constraints, the number of inde- pendent force field parameters is decreased. The same is true for the scaling factors: in the matrix Eq. (7) elements βij should be the same for all pairs of atoms that transfer to each other by the symmetry operations for the particu- lar molecule (that is, rotations and reflections that result in the molecular configuration identical to original). These constrains may be added to those defined by Eq. (7). Other model assumptions (e.g., equality of certain param- eters or negligence of the others) may also be accounted for when minimizing functional Eq. (3).
The general approach to account for molecular symme- try is based on the fact that direct problem of vibrational spectroscopy may be reduced to a series of independent problems, one for each symmetry type of the normal vi- brations. Symmetry coordinates for a molecule may be introduced as qs = Cq where q is a set of original coordi- nates (in this case, 3 N Cartesian coordinates), qs is a set of 3 N symmetry coordinates, and C is a known orthogo- nal matrix. In symmetry coordinates, force constant ma- trix Fs = CFCT is a block-diagonal matrix consisting of blocks corresponding to each symmetry type.
Technically, the described procedure may be reduced to a procedure dealing only with the symmetry block matrices Fs to minimize calculation complexity.
Another common option frequently used for adjustment of theoretical force constants is the usage of isotopic species of a molecule, thus increasing the set of experimentally available data. This option may also be applied to the case of Cartesian force field scaling. This procedure is also compatible with the above-described symmetry reduction if isotopic species of a molecule possess the same symmetry.
After completing minimization and finding scale matrix B, the force constant matrix in Cartesian coordinates is obtained from Eq. (2). It is then possible to convert it to any appropriate coordinate system for the further analysis; this transform will preserve adjusted vibrational frequencies.
Below, we demonstrate results of scaling force constants in Cartesian coordinates for the indole and pyrrole molecules, taking into account for both symmetry and additional experi- mental data of the deuterated molecules. These molecules were chosen as testing examples because (a) similar fragments exist in many biological systems; (b) both structure and vibra- tional spectra of indole and pyrrole are well investigated; and
(c) there exist good publications on the determination of scal- ing factors of both molecules in internal coordinates by the conventional Pulay method which are used for the compara- tive discussion. So, we have repeated the calculations of scaled force fields of these two molecules at the same theoret- ical level (B3LYP/6-31G**) with the same sets of experimen- tal data as in Refs. [22, 23] but in the Cartesian coordinates.
Computational details
Quantum mechanical calculations of indole and pyrrole molecules w ere p erformed with the p rogram GAUSSIAN 03 (Revision B.03) package [24]. For this molecule, we have performed the B3LYP [25] calculation with the 6-31G(d,p) basis. The minima of the potential surface were found by relaxing the geometric parameters with the standard optimization methods. Analytical force constants were derived and harmonic vibrational frequen- cies were calculated. Using theoretical force constants as F0 and sets of experimental frequencies collected in [22, 23] for indole and pyrrole, respectively, the inverse scal- ing problem was solved and scale factors were found as the extremal of functional Eq. (2). Calculation of scale factors was carried out with the special routine of the software package SPECTRUM [26] which has been sup- plied with the additional options mentioned in the previ- ous section.
The problem Eq. (1) with a set of constraints which were used in numerical calculations of scaling factors in Cartesian coordinates for the considered molecules proved to be incom- patible (explained by the incompatibility of experimental data for isotopic species within harmonic model);
Numerical applications
Indole molecule
Sixteen atomic indole molecule represents a compound with low symmetry (Cs). In this case, the whole Cartesian matrix has dimensions of 3 N = 48, while there exist 42 vibrational frequencies. Cartesian force matrix splits into blocks A′ and A″ (corresponding to in-plane and out-of- plane vibrations) with dimensions 32 × 32 and 16 × 16, respectively; these blocks contain 29 and 13 vibrational frequencies (Fig. 1).
In this case, a common set of scale factors was obtained that satisfies Eqs. (6)–(7). This set is shown in Table 1.
Table 2 shows quantum-chemical (B3LYP/6-31G(d,p)), experimental and fitted frequencies of the indole molecule.
Mean square error in frequency fitting is equal to 3.7 cm−1. Note that the largest difference between fitted and observed frequency (15 cm−1) observed for ν = 400 cm−1 (A″) is similar to deviation detected in where a standard Pulay scheme was applied. This discrep- ancy was attributed to high anharmonicity of this vibration mode and this frequency was excluded from fitting in [21]. To analyze the quality of fitting procedure, the ab initio and fitted Hessians of pyrrole were transformed to the set of redundant internal coordinates. In Tables S2 and S3, one can see the transformed to these internal coordi- nates matrices of force constants before and after scaling. It is evident that in general, the structure of matrix F did not change after fitting.
Pyrrole molecule
In this section, we present the examples of calculated scaling Cartesian force field factors for the molecule of pyrrole C4H4NH (Fig. 2).
A new specific feature in the application of the algorithm is the usage of the isotopic species of the molecule. We have used three experimental sets of frequencies [22] for completely and partially deuterated pyrrole (1-deuteropyrrole and pentadeuteropyrrole), all species possessing the same C2v symmetry.
Pyrrole molecule has N = 10 atoms and therefore 3 N-6 = 24 normal vibrations.
At the same time, 30 Cartesian coordinates of a molecule split into symmetry blocks as 10A1 þ 4A2 þ 10B1 þ 6B2
This means that each block contains one or more de- pendent coordinates. Coefficients in the last formula de- fine dimensions of the force constant matrices in Cartesian coordinates and corresponding scale factor ma- trices for each type of symmetry.
The experimental, theoretical, and fitted vibrational fre- quencies for isotopic species of pyrrole are presented in Table 3. In the calculations, we applied standard proce- dure Eqs. (6)–(7) to obtain a common set of scale factors as suggested in [15, 16]; these results are marked as BScaled I^. For comparison, we also present the results of individual scaling in each symmetry block (see Eq. (8)) that provides much better fitting of experimental frequencies (denoted as BScaled II^ in Table 3). The Cartesian force matrix of pyrrole scaled by the symmetry blocks is presented in Table S1.
Table 4 contains resulting matrix of βij coefficients corre- sponding to BScaled I^ results. The values of scaling factors presented in Table 4 reflect the symmetry C2v of a molecule, e.g., the diagonal values for carbon atom are equal for pairs of atoms: C1 and C4 (0.9843) and C2 and C3 (0.9775), similar in- pair equalities are observed in other cases. For the second ver- sion of scaling, we present Cartesian force matrix (Table 5). In the last line of Table 3, average fitting error for all fre- quencies is shown.
Discussion and summary
Analysis of the Tables 2 and 3 shows satisfactory correspon- dence between experimental and fitted frequencies compara- ble to the correspondence obtained with the use of conven- tional Pulay scaling in internal coordinates [22, 23]. This dem- onstrates that the model of correcting theoretical vibrational frequencies by means of scaling Cartesian force matrices ap- pears reasonable.
The diagonal scale factors for similar atoms in indole and pyrrole molecules appear quite close. The same is true for the pairs of bonded atoms (C, H); this shows good possibilities of transferring scale factors for the atoms in similar environment. Optimized values of factor βij are the same for all pairs of atoms that transfer to each other by the symmetry operations for the particular molecule. Note that it is also possible to apply the procedure for each symmetry block individually, yielding somewhat better frequency fitting, similar to what is often done for the standard scaling approach.
A new numerical algorithm for the calculation of scale factors for the molecular force fields expressed in Cartesian coordinates allows to reduce the difficulties related to the choice of internal coordinates in the com- plex molecules. The suggested method appears benefi- cial for calculating vibrational spectra of the large bio- logical molecules, associates, polymers, and nanostruc- tures where number of atoms exceeds hundreds and thousands, while only moderately accurate quantum- chemistry methods may be applied.
Acknowledgements This work was partially supported by the Russian Foundation for Basic Research grant.

(R)-N-(tert-Butoxycarbonyl)-3-hydroxypyrrolidineCatalog No.:AA003C4D CAS No.:109431-87-0 MDL No.:MFCD01317838 MF:C9H17NO3 MW:187.2362 |
1,5:2,3-Dianhydro-4,6-O-benzylidene-D-allitolCatalog No.:AA007ATZ CAS No.:109428-30-0 MDL No.:MFCD06657645 MF:C13H14O4 MW:234.2479 |
N-Butyl 4-chloropicolinamideCatalog No.:AA0082D3 CAS No.:1094306-27-0 MDL No.:MFCD11177050 MF:C10H13ClN2O MW:212.6760 |
(2S,3aS,6aS)-Octahydrocyclopenta[b]pyrrole-2-carboxylic acidCatalog No.:AA008RAL CAS No.:109428-53-7 MDL No.:MFCD03265236 MF:C8H13NO2 MW:155.1943 |
TERBIUM TRIS(HEXAMETHYLDISILAZIDE)Catalog No.:AA008YIP CAS No.:109433-86-5 MDL No.:MFCD03427092 MF:C18H54N3Si6Tb MW:640.0798 |
(2-Chloropyridin-3-yl)(4-(hydroxymethyl)piperidin-1-yl)methanoneCatalog No.:AA008ZMA CAS No.:1094301-19-5 MDL No.:MFCD11133074 MF:C12H15ClN2O2 MW:254.7127 |
4-[3-(Chloromethyl)phenyl]carbonylmorpholineCatalog No.:AA00HBEI CAS No.:1094300-44-3 MDL No.:MFCD11208347 MF:C12H14ClNO2 MW:239.6981 |
1-propanoyl-2,3-dihydro-1H-indole-5-carboxylic acidCatalog No.:AA00J5RB CAS No.:1094303-82-8 MDL No.:MFCD11177687 MF:C12H13NO3 MW:219.2365 |
1-(2-chloroethoxy)-2-methylcyclohexaneCatalog No.:AA018RTP CAS No.:1094273-74-1 MDL No.:MFCD11164766 MF:C9H17ClO MW:176.6837 |
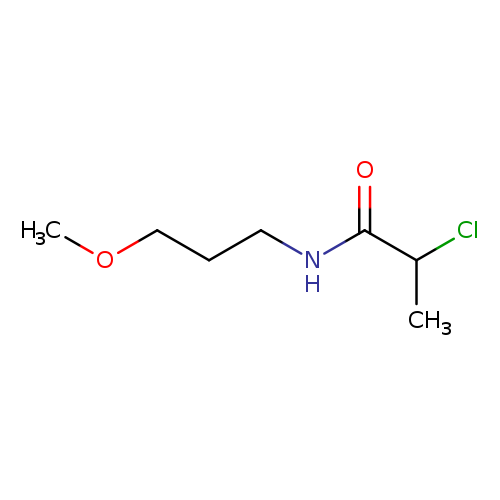
2-Chloro-N-(3-methoxypropyl)propanamideCatalog No.:AA019KMO CAS No.:1094292-31-5 MDL No.:MFCD11205017 MF:C7H14ClNO2 MW:179.6446 |
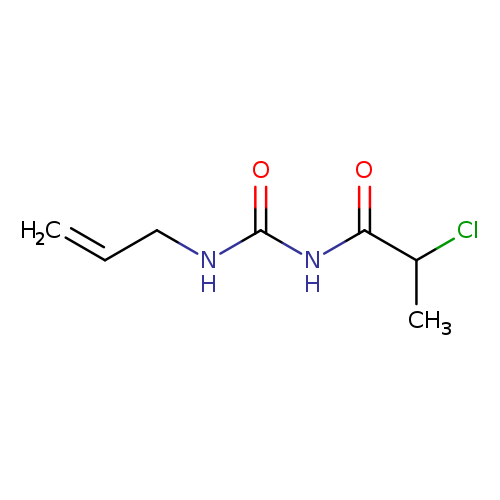
1-(2-chloropropanoyl)-3-(prop-2-en-1-yl)ureaCatalog No.:AA019KXK CAS No.:1094302-21-2 MDL No.:MFCD11185278 MF:C7H11ClN2O2 MW:190.6274 |
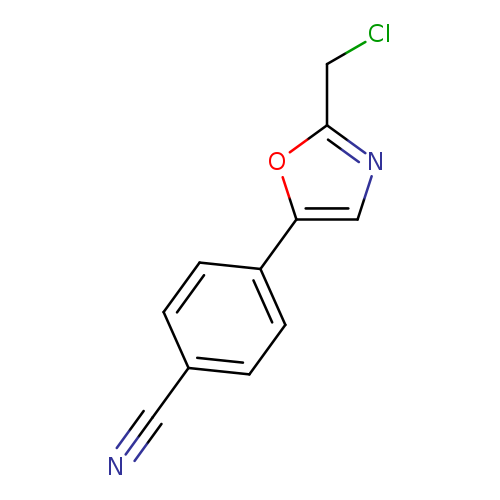
4-[2-(chloromethyl)-1,3-oxazol-5-yl]benzonitrileCatalog No.:AA019QWK CAS No.:1094318-26-9 MDL No.:MFCD11182417 MF:C11H7ClN2O MW:218.6391 |
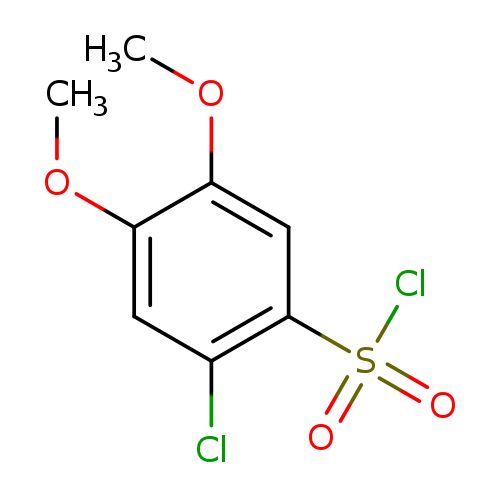
2-Chloro-4,5-dimethoxybenzenesulfonyl chlorideCatalog No.:AA019RAO CAS No.:1094292-46-2 MDL No.:MFCD13438190 MF:C8H8Cl2O4S MW:271.1177 |
4-Fluoro-3-[(1,2,4-oxadiazol-3-ylmethane)sulfonyl]benzoic acidCatalog No.:AA019RXX CAS No.:1094326-63-2 MDL No.:MFCD12633225 MF:C10H7FN2O5S MW:286.2364 |
1-methyl-3-(propan-2-yl)-1H-thieno[2,3-c]pyrazole-5-carboxylic acidCatalog No.:AA019SC3 CAS No.:1094271-45-0 MDL No.:MFCD11185460 MF:C10H12N2O2S MW:224.2795 |
4-(3-fluorophenyl)oxan-4-amineCatalog No.:AA019SPA CAS No.:1094283-08-5 MDL No.:MFCD11186129 MF:C11H14FNO MW:195.2334 |
1-benzyl-N-propylpiperidin-3-amineCatalog No.:AA019VFW CAS No.:1094325-69-5 MDL No.:MFCD11183086 MF:C15H24N2 MW:232.3645 |
2-bromo-6-(4-fluorophenyl)imidazo[2,1-b][1,3,4]thiadiazoleCatalog No.:AA019VRB CAS No.:1094275-22-5 MDL No.:MFCD11209935 MF:C10H5BrFN3S MW:298.1342 |
2-[2-(4-bromothiophen-2-yl)-1,3-thiazol-4-yl]acetic acidCatalog No.:AA019W01 CAS No.:1094293-78-3 MDL No.:MFCD11179467 MF:C9H6BrNO2S2 MW:304.1834 |
4-(4-bromophenyl)oxan-4-amineCatalog No.:AA019W3G CAS No.:1094283-04-1 MDL No.:MFCD11186108 MF:C11H14BrNO MW:256.1390 |
5-(chloromethyl)-1-[4-(trifluoromethyl)phenyl]-1H-1,2,3,4-tetrazoleCatalog No.:AA019W47 CAS No.:1094265-93-6 MDL No.:MFCD11134247 MF:C9H6ClF3N4 MW:262.6189 |
3-(chloromethyl)-N-(furan-2-ylmethyl)benzamideCatalog No.:AA019WBN CAS No.:1094300-46-5 MDL No.:MFCD11208352 MF:C13H12ClNO2 MW:249.6929 |
5-(chloromethyl)-1-(2,6-dimethylphenyl)-1H-1,2,3,4-tetrazoleCatalog No.:AA019WGL CAS No.:1094328-52-5 MDL No.:MFCD11134282 MF:C10H11ClN4 MW:222.6741 |
2-[4-(difluoromethoxy)phenyl]oxiraneCatalog No.:AA019WHI CAS No.:1094302-44-9 MDL No.:MFCD11185349 MF:C9H8F2O2 MW:186.1554 |
3-(3-aminophenoxy)propanamideCatalog No.:AA019WHA CAS No.:1094322-85-6 MDL No.:MFCD11206020 MF:C9H12N2O2 MW:180.2038 |
1-(1-Bromoethyl)-2,4-difluorobenzeneCatalog No.:AA019WHN CAS No.:1094272-78-2 MDL No.:MFCD11180314 MF:C8H7BrF2 MW:221.0420 |
3-ethyl-1-hydroxycyclopentane-1-carboxylic acidCatalog No.:AA019WV8 CAS No.:1094307-11-5 MDL No.:MFCD11181324 MF:C8H14O3 MW:158.1950 |
4-(3-amino-1,2,4-triazin-5-yl)benzonitrileCatalog No.:AA019WXJ CAS No.:1094296-78-2 MDL No.:MFCD11134074 MF:C10H7N5 MW:197.1961 |
1-N-(Cyclopropylmethyl)benzene-1,3-diamineCatalog No.:AA019WYJ CAS No.:1094324-01-2 MDL No.:MFCD11132711 MF:C10H14N2 MW:162.2316 |
1-[5-(2-iodophenyl)thiophen-2-yl]-2-methylpropan-1-oneCatalog No.:AA019WYK CAS No.:1094282-10-6 MDL No.:MFCD11207003 MF:C14H13IOS MW:356.2219 |
5-[4-(difluoromethoxy)phenyl]-1,2,4-triazin-3-amineCatalog No.:AA019X3I CAS No.:1094271-28-9 MDL No.:MFCD11185356 MF:C10H8F2N4O MW:238.1935 |
5-(2,5-Difluorophenyl)-1,2,4-triazin-3-amineCatalog No.:AA019XX6 CAS No.:1094296-76-0 MDL No.:MFCD11134069 MF:C9H6F2N4 MW:208.1675 |
5-(2-fluorophenyl)-1,2,4-triazin-3-amineCatalog No.:AA019Y1L CAS No.:1094296-86-2 MDL No.:MFCD11134109 MF:C9H7FN4 MW:190.1771 |
3-(Thiomorpholine-4-sulfonyl)thiophene-2-carboxylic acidCatalog No.:AA019Y2X CAS No.:1094283-18-7 MDL No.:MFCD11186167 MF:C9H11NO4S3 MW:293.3829 |
5-Chloro-2-(cyclopentyloxy)benzoic acidCatalog No.:AA019Y8D CAS No.:1094310-78-7 MDL No.:MFCD11180628 MF:C12H13ClO3 MW:240.6828 |
5-(1-chloroethyl)-3-[(4-fluorophenyl)methyl]-1,2,4-oxadiazoleCatalog No.:AA019Z0Q CAS No.:1094291-16-3 MDL No.:MFCD11205895 MF:C11H10ClFN2O MW:240.6613 |
7-chloro-3-methyl-[1,2,4]triazolo[4,3-c]pyrimidineCatalog No.:AA01A0DE CAS No.:1094292-91-7 MDL No.:MFCD11182612 MF:C6H5ClN4 MW:168.5837 |
2-(1-chloroethyl)-5-(3,4-dichlorophenyl)-1,3-oxazoleCatalog No.:AA01A22B CAS No.:1094318-44-1 MDL No.:MFCD11182485 MF:C11H8Cl3NO MW:276.5463 |
2-(1-Methyl-5-oxo-4,5-dihydro-1H-pyrazol-3-yl)acetic acidCatalog No.:AA01A28V CAS No.:1094269-98-3 MDL No.:MFCD11133565 MF:C6H8N2O3 MW:156.1393 |
N'-hydroxy-3-(2-methylmorpholin-4-yl)-3-oxopropanimidamideCatalog No.:AA01A2AW CAS No.:1094299-57-6 MDL No.:MFCD11208240 MF:C8H15N3O3 MW:201.2230 |
4-(2-chloroethyl)-2-methyl-3,4-dihydro-2H-1,4-benzoxazin-3-oneCatalog No.:AA01A30G CAS No.:1094288-54-6 MDL No.:MFCD11207473 MF:C11H12ClNO2 MW:225.6715 |
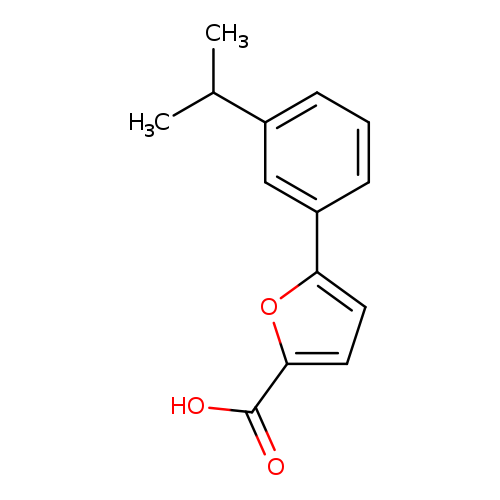
5-[3-(Propan-2-yl)phenyl]furan-2-carboxylic acidCatalog No.:AA01A4BF CAS No.:1094267-53-4 MDL No.:MFCD11208133 MF:C14H14O3 MW:230.2592 |
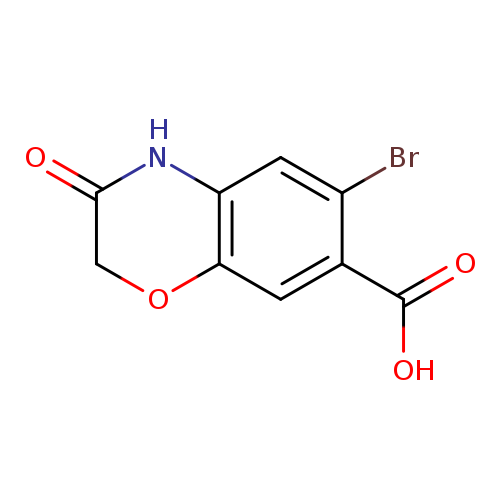
6-bromo-3-oxo-3,4-dihydro-2H-1,4-benzoxazine-7-carboxylic acidCatalog No.:AA01A4QP CAS No.:1094298-69-7 MDL No.:MFCD11206335 MF:C9H6BrNO4 MW:272.0522 |
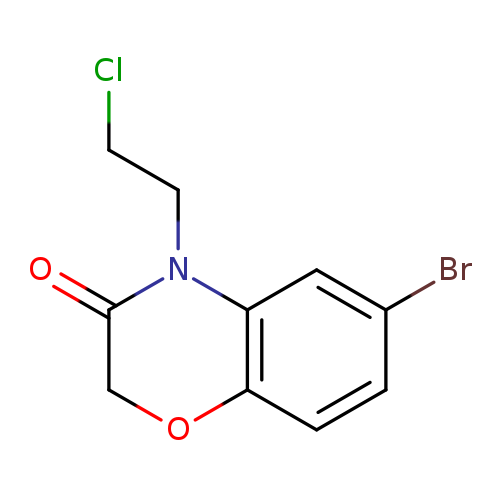
6-bromo-4-(2-chloroethyl)-3,4-dihydro-2H-1,4-benzoxazin-3-oneCatalog No.:AA01A55A CAS No.:1094298-71-1 MDL No.:MFCD11206341 MF:C10H9BrClNO2 MW:290.5410 |
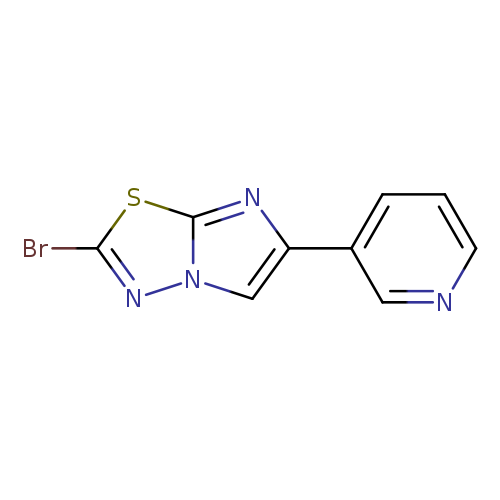
3-(2-Bromoimidazo[2,1-b][1,3,4]thiadiazol-6-yl)pyridineCatalog No.:AA01A64V CAS No.:1094279-90-9 MDL No.:MFCD11209938 MF:C9H5BrN4S MW:281.1318 |
3-tert-butyl-7-chloro-[1,2,4]triazolo[4,3-c]pyrimidineCatalog No.:AA01A6N0 CAS No.:1094292-89-3 MDL No.:MFCD11182605 MF:C9H11ClN4 MW:210.6634 |
1-[5-(4-chloro-2-fluorophenyl)thiophen-2-yl]ethan-1-oneCatalog No.:AA01A6T6 CAS No.:1094282-07-1 MDL No.:MFCD11206992 MF:C12H8ClFOS MW:254.7077 |
2-(pyridin-3-yloxy)benzoic acidCatalog No.:AA01A84O CAS No.:1094325-06-0 MDL No.:MFCD11182950 MF:C12H9NO3 MW:215.2048 |
5-bromo-7-chloro-3-methyl-1-benzofuran-2-carboxylic acidCatalog No.:AA01A8A3 CAS No.:1094288-67-1 MDL No.:MFCD11207508 MF:C10H6BrClO3 MW:289.5098 |
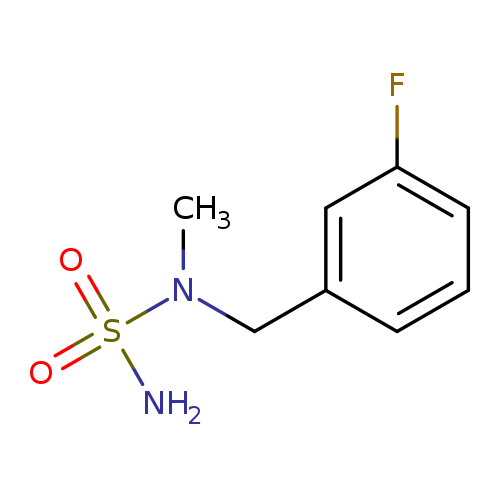
N-[(3-Fluorophenyl)methyl]-N-methylaminosulfonamideCatalog No.:AA01A8X1 CAS No.:1094315-37-3 MDL No.:MFCD11205213 MF:C8H11FN2O2S MW:218.2485 |
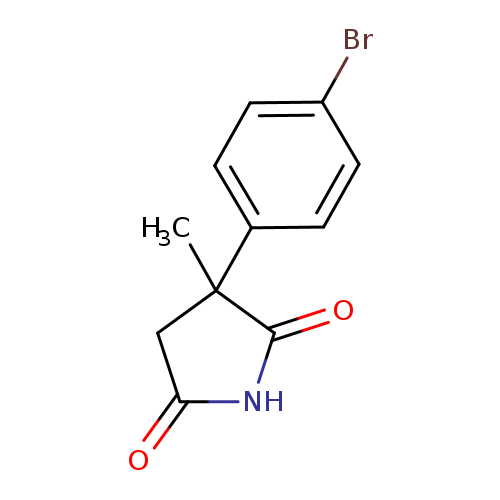
3-(4-bromophenyl)-3-methylpyrrolidine-2,5-dioneCatalog No.:AA01A9G3 CAS No.:1094289-44-7 MDL No.:MFCD11207821 MF:C11H10BrNO2 MW:268.1066 |
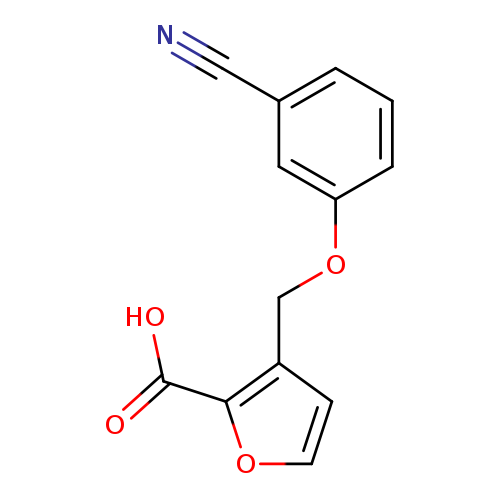
3-[(3-cyanophenoxy)methyl]furan-2-carboxylic acidCatalog No.:AA01A9GX CAS No.:1094298-96-0 MDL No.:MFCD11543928 MF:C13H9NO4 MW:243.2149 |
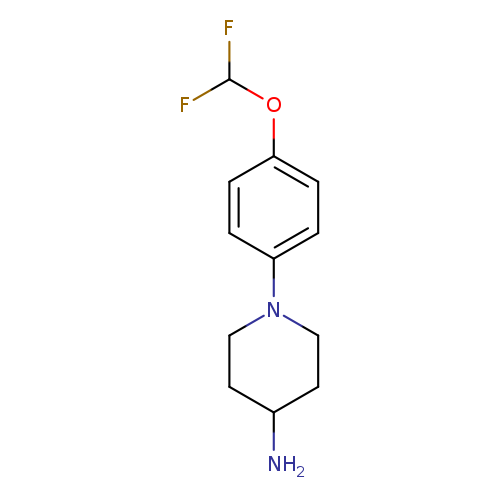
1-[4-(difluoromethoxy)phenyl]piperidin-4-amineCatalog No.:AA01A9IJ CAS No.:1094308-87-8 MDL No.:MFCD11185586 MF:C12H16F2N2O MW:242.2650 |
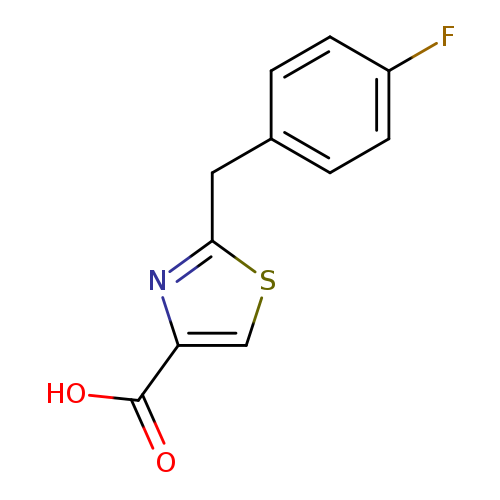
2-[(4-fluorophenyl)methyl]-1,3-thiazole-4-carboxylic acidCatalog No.:AA01A9K6 CAS No.:1094291-21-0 MDL No.:MFCD11205906 MF:C11H8FNO2S MW:237.2501 |
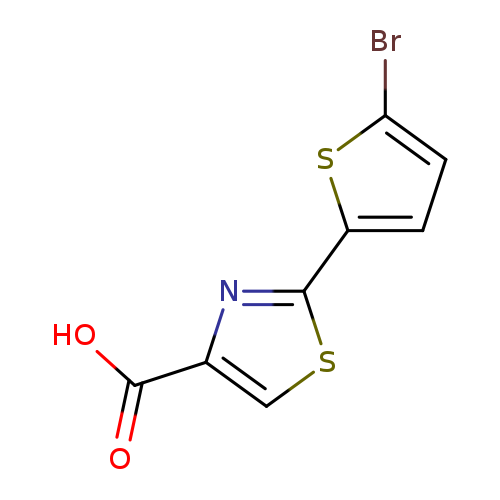
2-(5-bromothiophen-2-yl)-1,3-thiazole-4-carboxylic acidCatalog No.:AA01A9YK CAS No.:1094293-74-9 MDL No.:MFCD07376391 MF:C8H4BrNO2S2 MW:290.1569 |
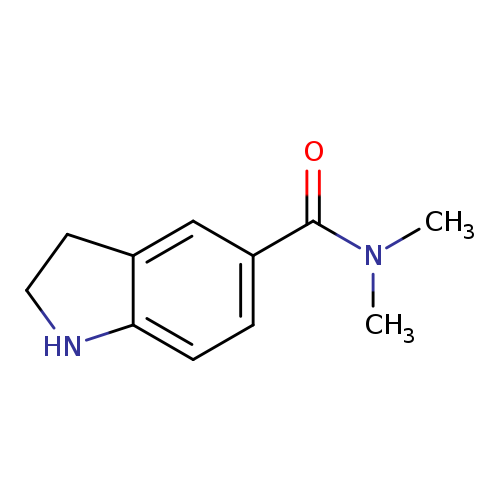
N,N-dimethyl-2,3-dihydro-1H-indole-5-carboxamideCatalog No.:AA01AA2P CAS No.:1094284-83-9 MDL No.:MFCD11181066 MF:C11H14N2O MW:190.2417 |
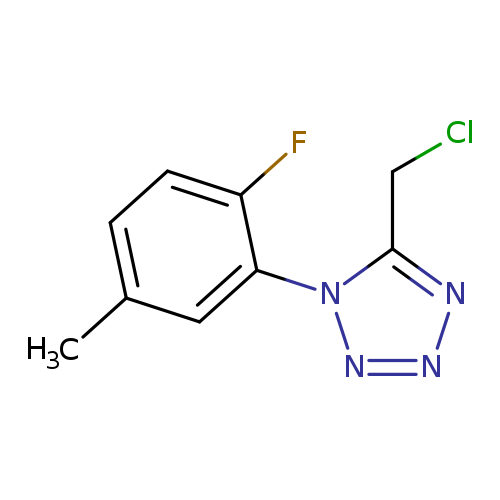
5-(chloromethyl)-1-(2-fluoro-5-methylphenyl)-1H-1,2,3,4-tetrazoleCatalog No.:AA01AAC5 CAS No.:1094328-54-7 MDL No.:MFCD11134291 MF:C9H8ClFN4 MW:226.6380 |
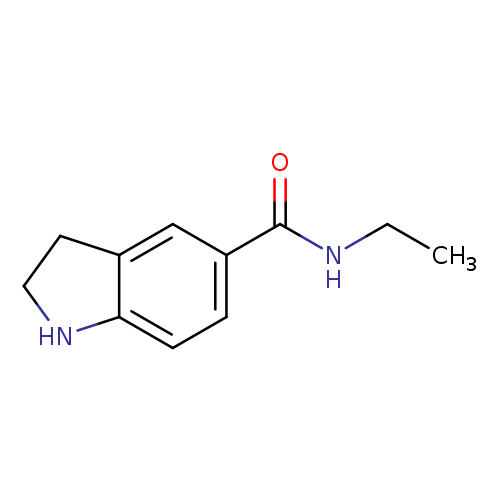
N-ethyl-2,3-dihydro-1H-indole-5-carboxamideCatalog No.:AA01AAP3 CAS No.:1094284-63-5 MDL No.:MFCD11181011 MF:C11H14N2O MW:190.2417 |
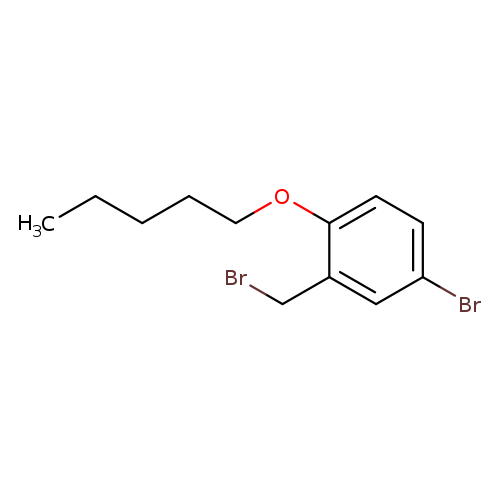
4-Bromo-2-(bromomethyl)-1-(pentyloxy)benzeneCatalog No.:AA01AAXU CAS No.:1094273-06-9 MDL No.:MFCD11180444 MF:C12H16Br2O MW:336.0628 |
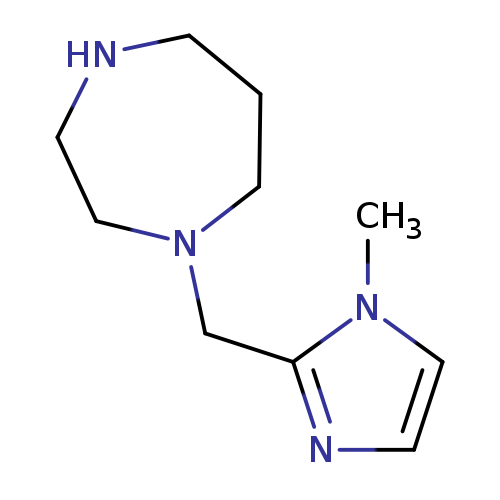
1-[(1-methyl-1H-imidazol-2-yl)methyl]-1,4-diazepaneCatalog No.:AA01AB8B CAS No.:1094317-96-0 MDL No.:MFCD11182271 MF:C10H18N4 MW:194.2767 |
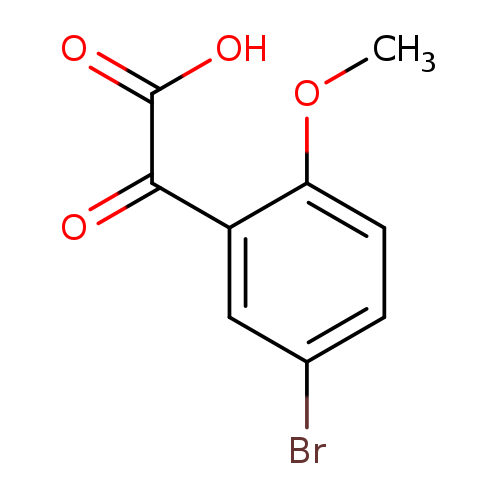
2-(5-bromo-2-methoxyphenyl)-2-oxoacetic acidCatalog No.:AA01ABAN CAS No.:1094294-15-1 MDL No.:MFCD11179623 MF:C9H7BrO4 MW:259.0535 |
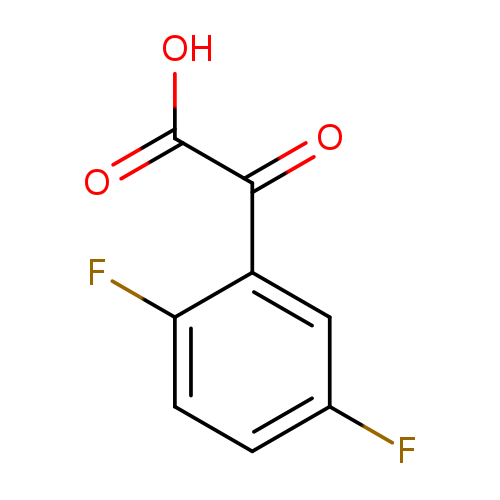
2-(2,5-DIFLUOROPHENYL)-2-OXOACETIC ACIDCatalog No.:AA01ACXN CAS No.:1094294-16-2 MDL No.:MFCD11179634 MF:C8H4F2O3 MW:186.1124 |
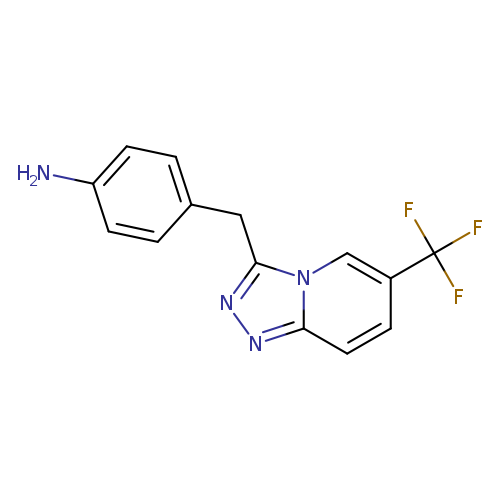
4-{[6-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl]methyl}anilineCatalog No.:AA01AD1H CAS No.:1094315-12-4 MDL No.:MFCD13467475 MF:C14H11F3N4 MW:292.2591 |
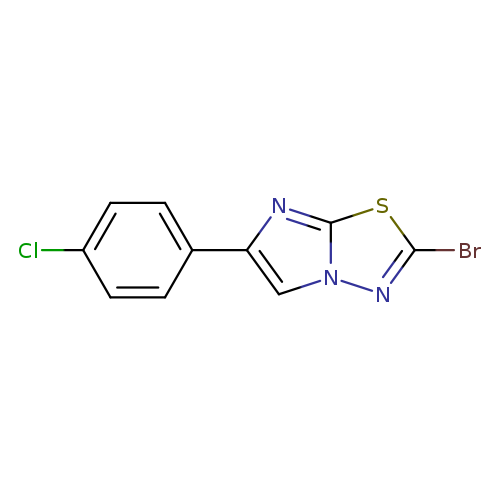
2-bromo-6-(4-chlorophenyl)imidazo[2,1-b][1,3,4]thiadiazoleCatalog No.:AA01AFU7 CAS No.:1094279-94-3 MDL No.:MFCD11209952 MF:C10H5BrClN3S MW:314.5888 |
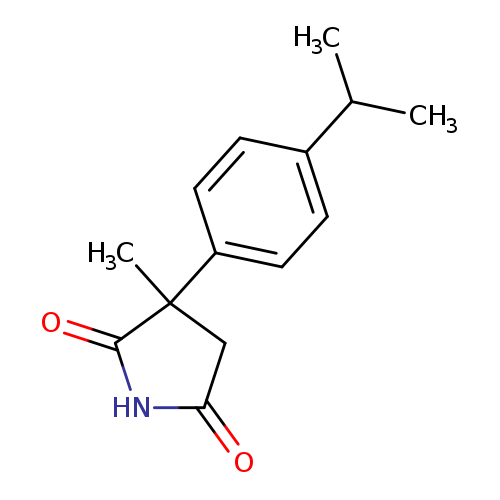
3-methyl-3-[4-(propan-2-yl)phenyl]pyrrolidine-2,5-dioneCatalog No.:AA01ALOT CAS No.:1094289-46-9 MDL No.:MFCD11207828 MF:C14H17NO2 MW:231.2903 |
4-({5H,6H,7H-pyrrolo[2,1-c][1,2,4]triazol-3-yl}methyl)anilineCatalog No.:AA01B13M CAS No.:1094285-37-6 MDL No.:MFCD11181265 MF:C12H14N4 MW:214.2664 |
2-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-6-yl)-2-oxoacetic acidCatalog No.:AA01B97T CAS No.:1094294-14-0 MDL No.:MFCD11179620 MF:C10H6N2O5 MW:234.1650 |
3-(2-ethoxyethyl)-1,2-oxazol-5-amineCatalog No.:AA01BA20 CAS No.:1094310-65-2 MDL No.:MFCD11178747 MF:C7H12N2O2 MW:156.1824 |
1-(Prop-2-enoyl)-1,2,3,4-tetrahydroquinoline-6-carboxylic acidCatalog No.:AA01BCFD CAS No.:1094284-48-6 MDL No.:MFCD11180951 MF:C13H13NO3 MW:231.2472 |
5-(2-Bromoacetyl)-1-methylindolin-2-oneCatalog No.:AA01BCJP CAS No.:1094318-21-4 MDL No.:MFCD11182382 MF:C11H10BrNO2 MW:268.1066 |
3-ethyl-3-(4-methylphenyl)oxolane-2,5-dioneCatalog No.:AA01BCUJ CAS No.:1094328-65-0 MDL No.:MFCD11134333 MF:C13H14O3 MW:218.2485 |
2-(2-[(Difluoromethyl)sulfanyl]-4-methyl-1,3-thiazol-5-yl)acetic acidCatalog No.:AA01BF67 CAS No.:1094316-49-0 MDL No.:MFCD11186370 MF:C7H7F2NO2S2 MW:239.2628 |
1-(2,5-difluorophenyl)-2-methylpropan-1-oneCatalog No.:AA01BGY7 CAS No.:1094292-49-5 MDL No.:MFCD11205127 MF:C10H10F2O MW:184.1826 |
3-cyclopropyl-3-oxo-2-phenylpropanenitrileCatalog No.:AA01BP0Y CAS No.:1094320-77-0 MDL No.:MFCD11208543 MF:C12H11NO MW:185.2218 |
5-(cyanomethanesulfonyl)-2-fluorobenzoic acidCatalog No.:AA01BTBW CAS No.:1094326-59-6 MDL No.:MFCD11179904 MF:C9H6FNO4S MW:243.2116 |
2-Methyl-3-oxo-3-(pyridin-4-yl)propanenitrileCatalog No.:AA01BTL9 CAS No.:1094267-19-2 MDL No.:MFCD11208746 MF:C9H8N2O MW:160.1726 |
(4-Chloro-2-pyridinyl)-4-morpholinylmethanoneCatalog No.:AA01BU8P CAS No.:1094306-26-9 MDL No.:MFCD11177047 MF:C10H11ClN2O2 MW:226.6595 |
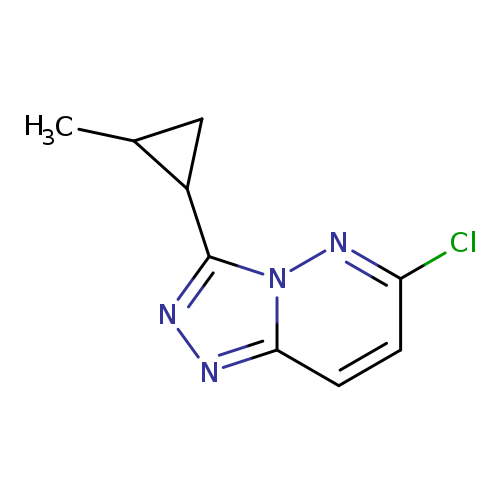
6-Chloro-3-(2-methylcyclopropyl)-[1,2,4]triazolo[4,3-b]pyridazineCatalog No.:AA01C1TC CAS No.:1094292-73-5 MDL No.:MFCD11182551 MF:C9H9ClN4 MW:208.6476 |
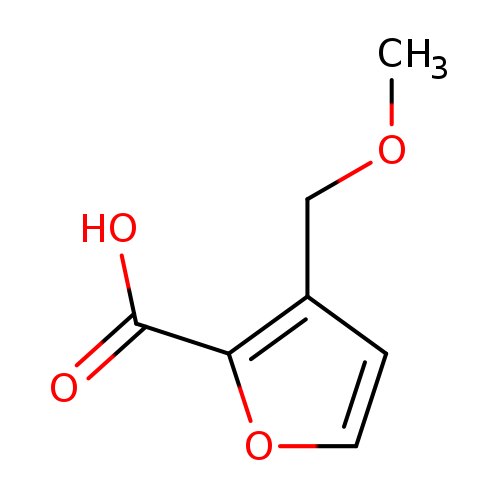
3-(Methoxymethyl)furan-2-carboxylic acidCatalog No.:AA01C79Y CAS No.:1094269-22-3 MDL No.:MFCD11206422 MF:C7H8O4 MW:156.136 |
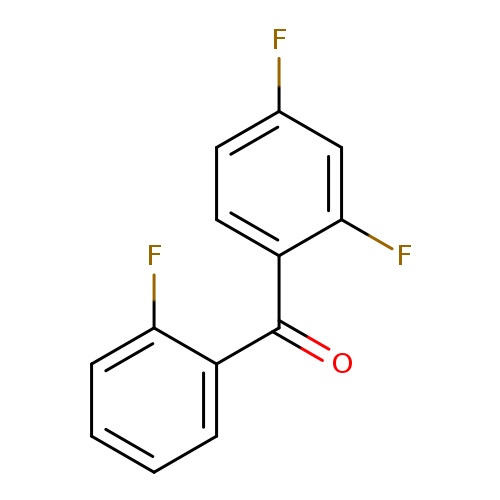
(2,4-difluorophenyl)(2-fluorophenyl)methanoneCatalog No.:AA01C996 CAS No.:1094311-98-4 MDL No.:MFCD11210396 MF:C13H7F3O MW:236.1893 |
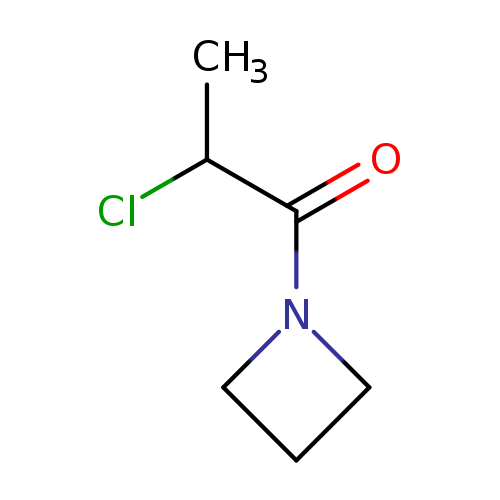
1-(azetidin-1-yl)-2-chloropropan-1-oneCatalog No.:AA01DUW3 CAS No.:1094294-06-0 MDL No.:MFCD11179592 MF:C6H10ClNO MW:147.6027 |
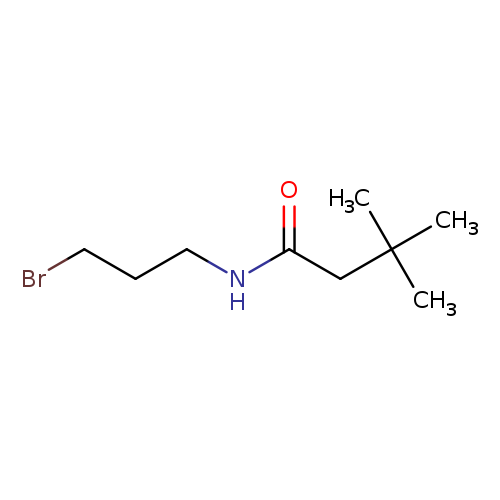
N-(3-bromopropyl)-3,3-dimethylbutanamideCatalog No.:AA01DUW4 CAS No.:1094301-76-4 MDL No.:MFCD11133320 MF:C9H18BrNO MW:236.1493 |
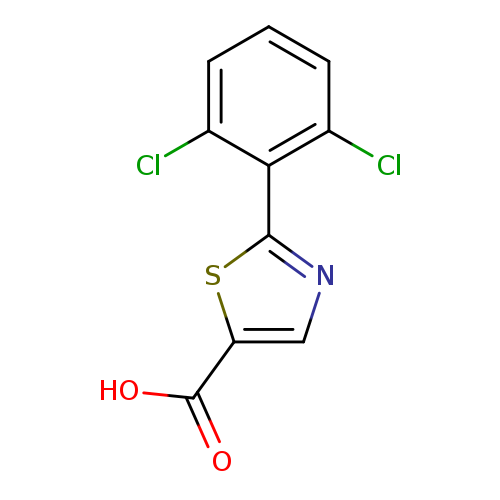
2-(2,6-dichlorophenyl)-1,3-thiazole-5-carboxylic acidCatalog No.:AA01E81X CAS No.:1094320-68-9 MDL No.:MFCD11208442 MF:C10H5Cl2NO2S MW:274.1232 |
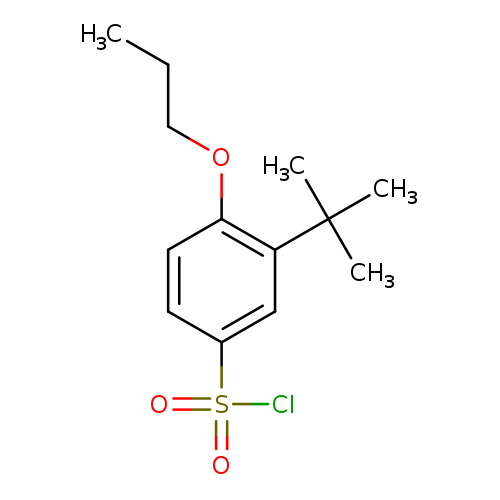
3-tert-butyl-4-propoxybenzene-1-sulfonyl chlorideCatalog No.:AA01E8OE CAS No.:1094296-52-2 MDL No.:MFCD11133934 MF:C13H19ClO3S MW:290.8062 |
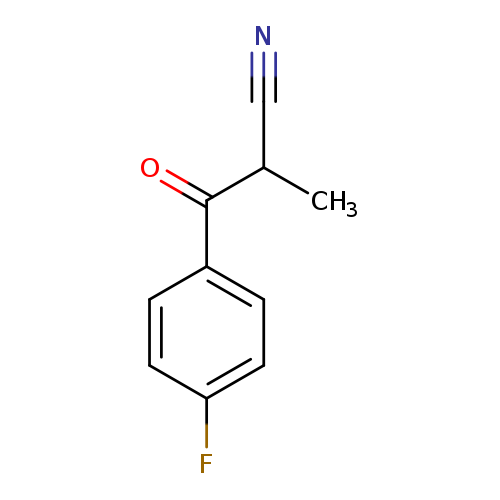
3-(4-fluorophenyl)-2-methyl-3-oxopropanenitrileCatalog No.:AA01EAVV CAS No.:1094267-29-4 MDL No.:MFCD11208803 MF:C10H8FNO MW:177.1750 |
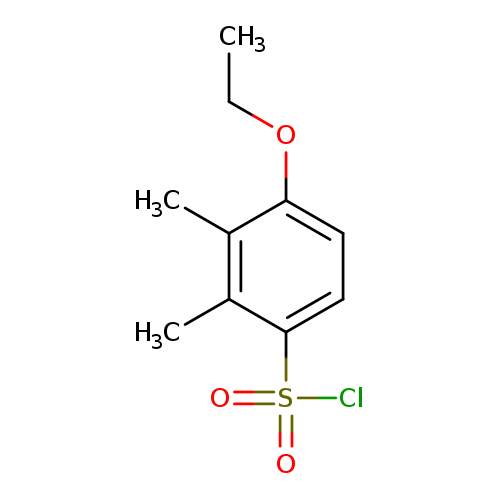
4-ethoxy-2,3-dimethylbenzene-1-sulfonyl chlorideCatalog No.:AA01EIRR CAS No.:1094296-27-1 MDL No.:MFCD11133871 MF:C10H13ClO3S MW:248.7264 |
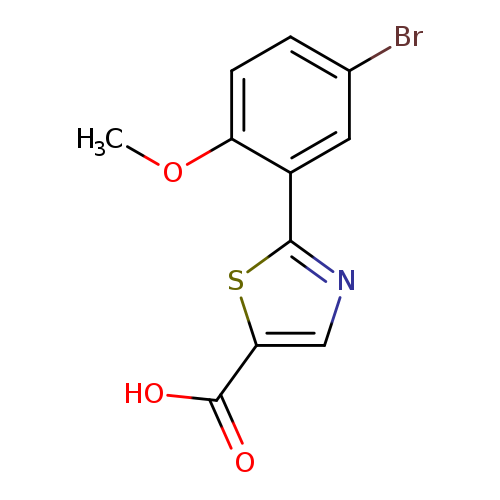
2-(5-bromo-2-methoxyphenyl)-1,3-thiazole-5-carboxylic acidCatalog No.:AA01EL66 CAS No.:1094320-72-5 MDL No.:MFCD11208450 MF:C11H8BrNO3S MW:314.1551 |
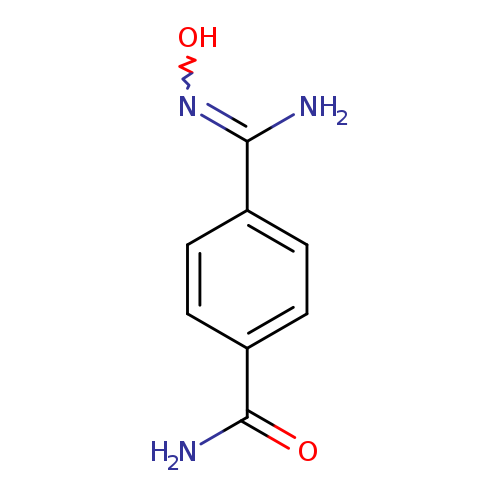
4-(N'-hydroxycarbamimidoyl)benzamideCatalog No.:AA01ELMH CAS No.:1094300-17-0 MDL No.:MFCD11208308 MF:C8H9N3O2 MW:179.176 |
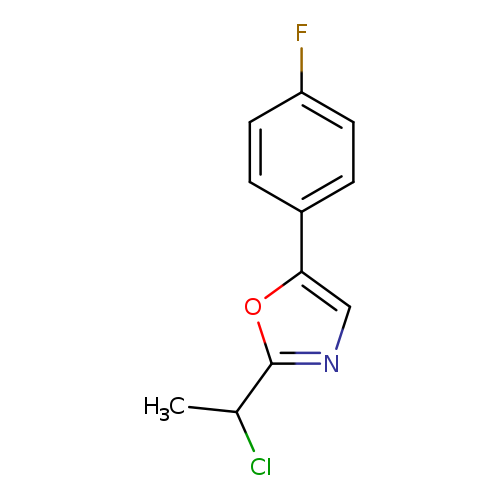
2-(1-chloroethyl)-5-(4-fluorophenyl)-1,3-oxazoleCatalog No.:AA01ELT6 CAS No.:1094318-38-3 MDL No.:MFCD11182462 MF:C11H9ClFNO MW:225.6467 |
4-[(4-chlorophenyl)methoxy]-3-nitrobenzaldehydeCatalog No.:AA01FOO2 CAS No.:1094305-11-9 MDL No.: MF:C14H10ClNO4 MW:291.6865 |
