2019-10-16 15:58:40
Recent Developments in the Chemistry of Boron Heterocycles
Brian J. Wang, Michael P. Groziak*
Department of Chemistry & Biochemistry, California State University East Bay, Hayward, CA, USA
*Corresponding author: E-mail: [email protected]
Contents
1.Introduction 48
2.Monoheteraboroles and Monoheteraborines 49
3.Diheteraboroles and Diheteraborines 69
4.Triheteraboroles and Triheteraborines 81
5.Tetraheteraboroles and Tetraheteraborines 82
6.Borazines and Boroxins 84
7.Concluding Remarks 86
Acknowledgments 86
References 86
Abstract
The utility of boron-containing compounds extends beyond the well-known applica- tions of boronic acids and esters in Suzuki cross-couplings and enzyme inhibition. In this selective review, the last 15 years’ worth of advances in the synthesis and utility of boron heterocycles are highlighted. The synthesis of isoelectronic analogs of benzene-based molecules and explorations into their physicochemical properties has been the primary research focus of several groups. Cyclocondensations of boronic acids, borate esters, boranes, or boron di-/trihalides with various functionalized reagents are a commonly explored route to the synthesis of complex structures, often with considerable regio- or stereoselectivity that is important to drug development. A more recent and perhaps lesser known approach to the synthesis of boron hetero- cycles involves photochemical methodologies. Often the highly p-conjugated systems that arise from this assortment of methodologies display an array of interesting optical properties that make them candidates in furthering the advancement of organic optical light-emitting devices.
Keywords: Borazines; Boron heterocycles; Boroxins; Cyclocondensation; Heteraborines; Heteraboroles; Isoelectronic; Isosteric; Macrocycles
Advances in Heterocyclic Chemistry, Volume 118
ISSN 0065-2725
http://dx.doi.org/10.1016/bs.aihch.2015.10.002
© 2016 Elsevier Inc.
All rights reserved.
1.INTRODUCTION
The utility of boron in heterocyclic chemistry extends well beyond the Suzuki cross-coupling reaction (1995CRV2457), which discards the boron atom of a boronic acid or ester under the basic conditions of this now famous Pd-catalyzed CeC bond-forming reaction. It is important for chemists to appreciate that boron is an element that can be valuable to retain in a molecule so that its unique properties can be utilized. One way of doing this is to embed it in a heterocycle ring that is designed to be stabilized by at least some degree of heteroaromaticity.
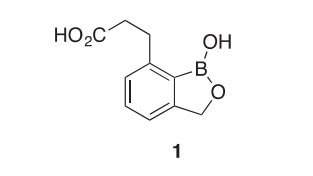
This contribution highlights the chemistry of many of the stable, formally aromatic boron heterocycles reported in the years 2000e2015. The chemical literature up through 1999 was covered by a similar review about 15 years ago (2000PHC1), and the most promising new boron compounds from a drug development perspective were also reviewed at that time (2001AJT321). The major goal of these two reviews was to convince the heterocyclic and pharmaceutical chemistry communities of the mostly untapped potential held by boron compounds. Unique structures and unusual reactivities can arise when this member of the second row of the periodic table is installed in a molecule in a deliberate and judicious manner. Much progress has been made. In the last decade, the FDA approved the first two boron-
containing drugs ever brought to market: Velcade® (bortezomib) for treating multiple myeloma (1998BMCL333) and Kerydin® (tavaborole) as a topical antifungal solution for treating onychomycosis (2006JMC4447).
Since the time of the aforementioned reviews, more recent advances in the chemistry of boron therapeutics have been covered by others (2009MI1275). Advances in the chemistry of azaborines have been reviewed recently (2012AG(I)6074), as have those in the medicinal chem- istry of boronic acid derivatives, 1,2-azaborines, and icosahedral boranes (carboranes) (2013AJC1118). Advances in the chemistry of tetrahedrally coordinated B-heterocycles have been reviewed (2000CCR85), and advances in luminescent dyes containing tetrahedral boron centers (excluding the well-known boron-dibenzopyrromethene (BODIPY) dyes and acetylacetonate boron complexes) have also been reviewed (2014AG(I)2290). Advances in the syntheses of boronenitrogen (B-N) for CeC bond replacement analogs of aromatic compounds, their physical properties, and applications have also been explored (2012CJC8, 2015CEJ3528). In the present review, the findings of certain select recent explorations into the synthesis, structures, reactivities, and physicochemical properties of the boron heterocycles in general are presented with the aim of stimulating the imagination of heterocyclic chemists working in a wide variety of subdisciplines.
2. MONOHETERABOROLES AND MONOHETERABORINES
A series of analogs of 3-(1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol- 7-yl)propanoic acid (1) has been synthesized and tested for activity against Plasmodium falciparum in an effort to explore this new benzoxaborole class of antimalarial agents (2011BMCL644). Compound 1 demonstrated the best potency (IC50 26 nM), supporting the premise that a carboxy- substituted side chain is required for potent antimalarial activity.
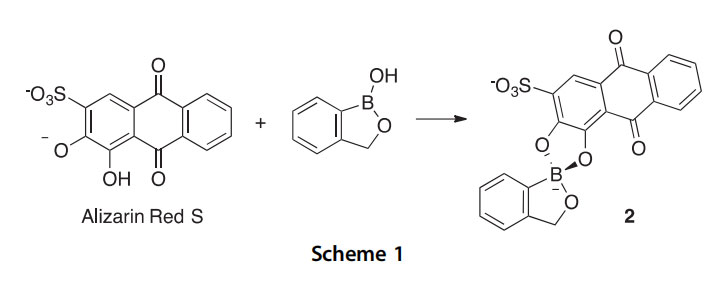
The reaction of benzoxaborole with alizarin red S, a catechol fluorescent dye, was studied kinetically over a range of pH values, revealing that condensation to form adduct 2 takes place with both the neutral boronic acid and anionic borate forms (Scheme 1) (2012JOC11200).
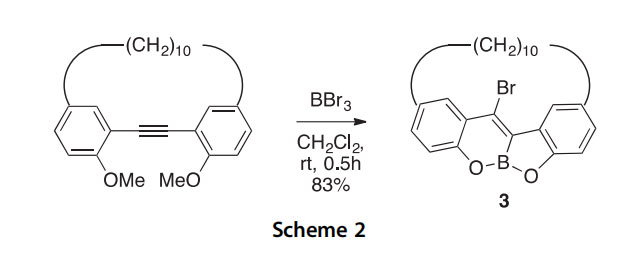
Synthesis and BBr3-mediated demethylation of 4,22-dimethoxy[2.10] metacyclophan-1-yne led to the novel [10](2,9)-5a,11a-benzofuro-5a- bora-11-bromochromenophane (3) in good yield (Scheme 2) (2012CJC441). A small set of intramolecularly chelated pinacol-protected 1-bora- isoindoles 4e7 were prepared to seek potential antimycobacterial agents against the Mycobacterium tuberculosis organism (2014HAC100). All were active (IC50 2.2e11.2 mg/ml), and the crystal structure of derivative 6 revealed only a slightly elongated N/B bond.
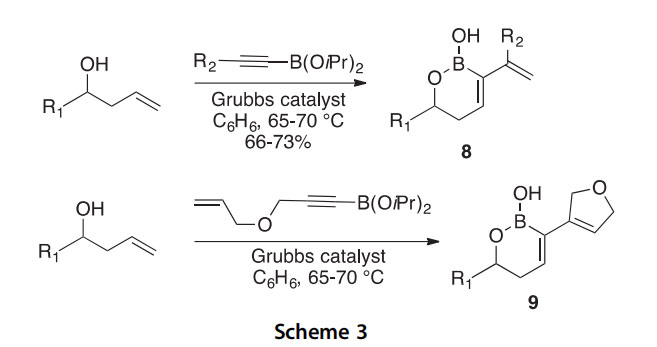
Dialkenylboronic acids 8 were synthesized in 66e73% yield by the ring closing ene-yne metathesis of alkynylboronic esters with homoallylic alco- hols in the presence of Grubbs’ catalyst (Scheme 3) (2002AG(I)3272). This also afforded bicyclic compounds 9 by combining the alkynylboronic ester annulation with an additional ring-closing metathesis (RCM), as demonstrated by an allyloxy-substituted alkynylboronic acid. An allenyla- tion reaction and an oxidation to a functionalized enone were demonstrated on the dialkenylboronic acids to emphasize the utility of this reaction in diversity oriented syntheses.
The synthesis of seven cyclic alkenyl boronic half acids (10a-g, 30e72% yield) has been accomplished via a RCM reaction using Grubbs’ 1st gener- ation catalyst (Scheme 4) (2010JOC6001).
A sequential three-step process consisting of diaryl ether lithiation, bor- ylation, and hydrolysis transforms dibenzofurans into dibenzoxaborininols 11 (Scheme 5) (2013MI3625)
Phenothiazinyldienes have been reduced by H3B$SMe2 to give the new fused ring tetrazolo[5,1-f ][1,2]azaborinin derivatives 12a-d (Scheme 6) (2010JOM2673). Besides extensive high-field 1D and 2D solution and solid-state multinuclear NMR characterization of 12a-d, X-ray crystallog- raphy of 12b was performed, revealing its zwitterionic character involving a bridge-head nitrogen atom.
The reaction between trimethylsilyl azide and pentaphenylborole has been investigated theoretically to suggest that it proceeds via azide to borole
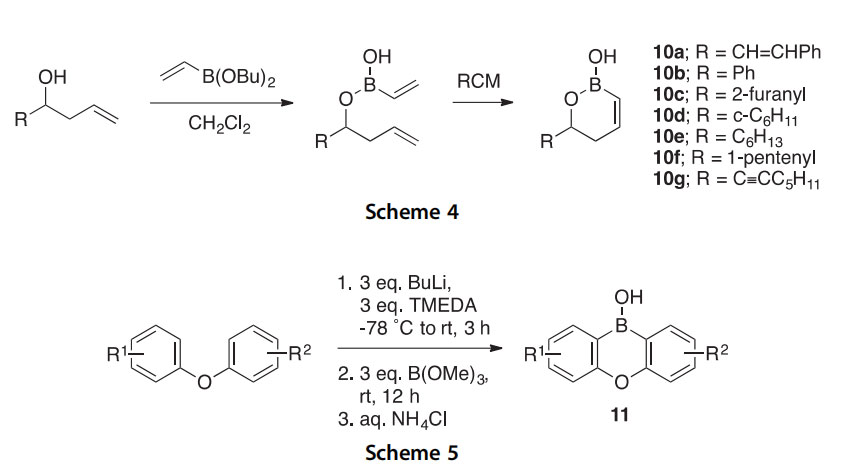
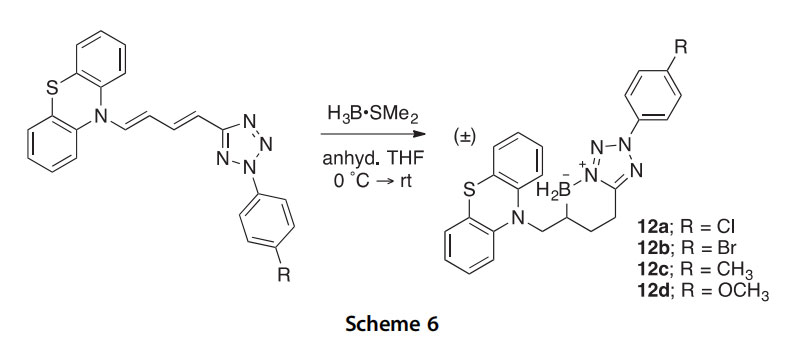
coordination, rearrangement to a bicyclic species, and conversion to the kinetic boron heterocycle product 13 or expulsion of N2 to the thermody- namic 1,2-azaborine product 14 (Scheme 7) (2014CC11724). The unusual cyclooctatetraene 13 was structurally characterized as a borole adduct. A sepa- rate investigation of the reactions of pentaphenylborole with 1-adamantyl- and 4-methoxyphenylisocyanate, and benzaldehyde and benzophenone produced the new seven-membered boron heterocycles 15, 16, and 17a,b, respectively, in high yields (2015IC1869).
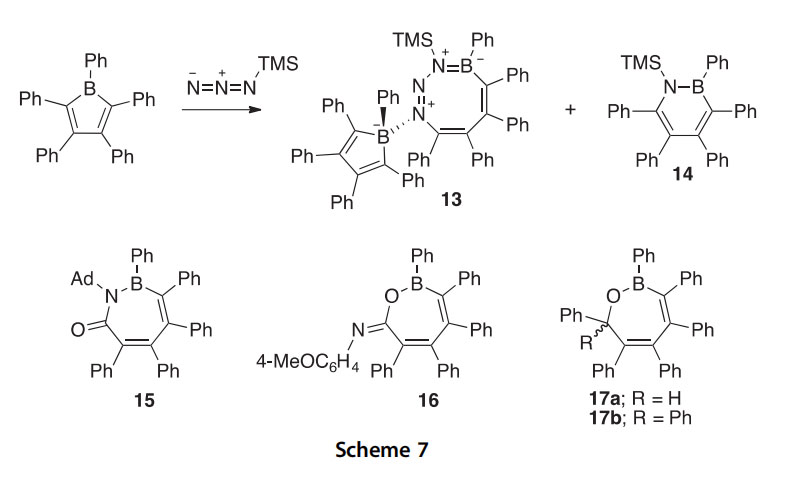
A catalytic route to 1,2-azaborinines has been developed using the [2 2]/[2 4] cycloaddition of di-tert-butyliminoborane and ethynylfer- rocene in the presence of a rhodium catalyst (Scheme 8) (2014AG(I) 3500). Thus, 1,2-azaborinine 18, the first ferrocene-functionalized boron heterocycle, was prepared and analyzed by X-ray crystallography.
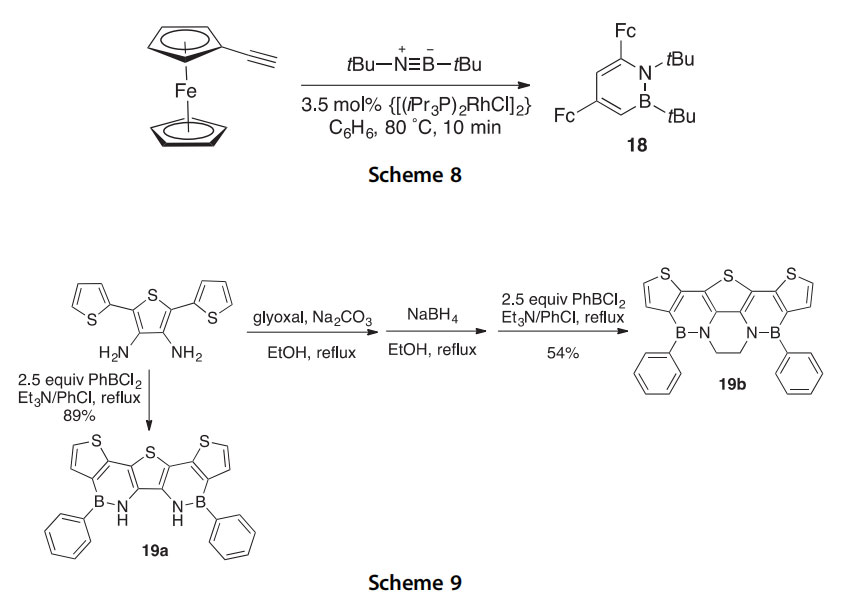
Novel azaborine and thiophene containing heteroacenes 19a,b were synthesized by the reaction of diaminoterthiophene derivatives with dichlorophenylborane in 54e89% yield (Scheme 9) (2010CC7007). X-ray crystallographic analysis of these heteroacenes revealed them to be mostly planar, with the azaborine moieties helping to rigidify the struc- tures, and also to exhibit p-stacking in the solid state. UVevis absorption spectra show that both compounds have red-shifted absorbance values when compared to similar reference compounds (lmax 395 nm and lmax 397 nm for 19a and 19b, respectively) and exhibit a deep-blue photoluminescence (lmax 407e410). These properties may lead to their potential use in semiconducting applications.
The B-N-fused coronene derivative 1,5,9-triaza-2,6,10-triphenylbora- coronene 20 and the N,N-dicyclohexyl-1-aza-12-borabenzoperylene diimide 21, both obtained via condensation of their respective anilines with PhBCl2 (Et3N in PhCH3, heat), have been investigated for photophys- ical properties that may advance new organic light-emitting diode (OLED) technology (2015OL560, 2015JOC196). Upon exposure to wet organic solvents, the former product (20) underwent OH for Ph group replace- ments, but the latter (21) did not. Boron heterocycle 21 proved to be a high- ly sensitive and selective fluoride anion sensor.
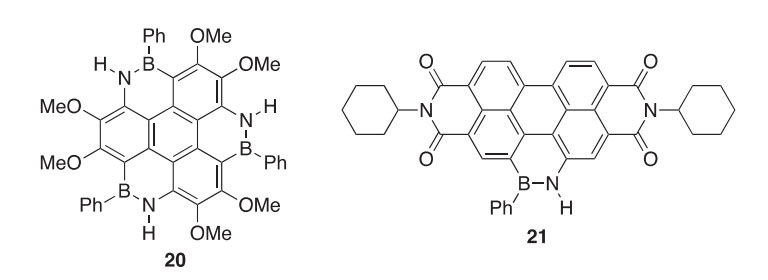
Several BN isosteres of perylene (22a-c), in which two CeC bonds are replaced by two isoelectronic BN bonds, have been synthesized in low iso- lated yields (3e17%) (2010CJC426). The presence of product 22a was inferred by a slight fluorescence of the isolated compound, but inadequate amounts of this product were isolated to conduct much meaningful charac- terization. In previous work, it was observed that functionalization of the pyridine ring with alkyl substituents served to buttress similar isosteres against air and moisture sensitivity. However, this buttressing effect was minimal in perylene isosteres as observed by irreversible one-electron reductions during cyclic voltammetry on 22a,b reflecting the low aromatic character of the central rings. X-ray crystallography of derivative 22b revealed the central ring system to be nearly planar.
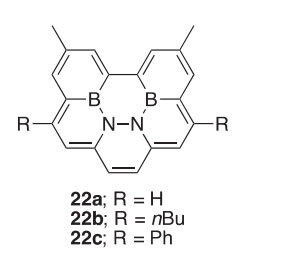
A macrocyclic dimer (23) containing an OBOBO structural unit has been prepared in 62% yield by the reaction of 2-aminophenol with 2- formylphenylboronic acid in anhydrous EtOH (Scheme 10) (2002CJC31). This dimer is also formed when a Schiff base containing boronate ester is treated with wet EtOH, implying that cleavage of the pinacol group generates the boronic acid before completing the cyclization. Analyses by X-ray crys- tallography as well as 11B NMR has confirmed that the dimer exists with coordinate covalent N/B interactions.
The B-chloromethylated version of benzo[e][1,2]azaborinine (2,1- borazaronaphthalene) 24 has been synthesized and found to be reactive
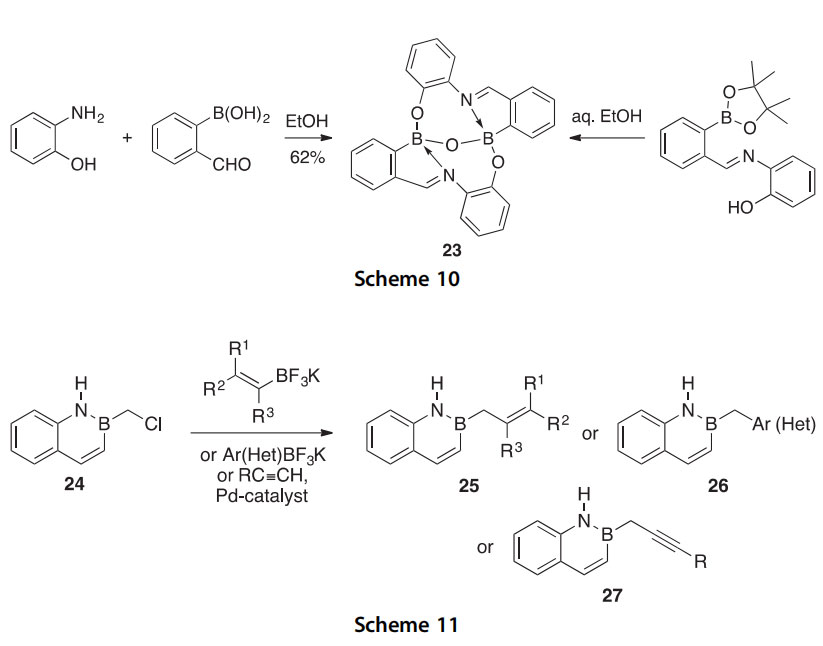
with nitrogen, oxygen, and sulfur nucleophiles (Scheme 11) (2014OL5636). It was shown to be a versatile building block by participating in Pd-catalyzed cross-coupling with potassium (hetero)aryltrifluoroborates, and alkenyltri- fluoroborates, and terminal alkynes (2014OL6024). Presented were 12 examples of products with general structure 25 (75e91% yields), 22 exam- ples of products with general structure 26 (45e92% yields), and 8 examples of products with general structure 27 (60e84% yields).
In related work, the B-arylated, C-brominated version of benzo[e][1,2] azaborinine 28 was shown to undergo self-arylation when treated with a Pd-catalyst in the presence of base, producing the 1,2-borazaro versions 29 of b-naphthol (Scheme 12) (2014JOC8339). Thirteen examples of prod- ucts with the general structure 29 (59e98% yields) were described. Substituted benzo[e][1,2]azaborinines like 28 were also found susceptible to Ni-catalyzed reductive cross-coupling reactions (2014OL3692).
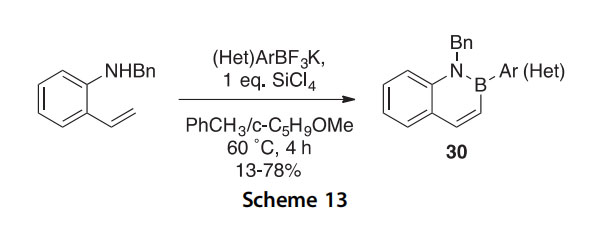
In addition, 2-aminostyrenes have been shown to condense with potas- sium organotrifluoroborates in the presence of the in situ fluorophile SiCl4 to give the functionalized benzo[e][1,2]azaborinines 30 (Scheme 13) (2014JOC365). Sixteen examples of products with this general structure (13e78% yields) were presented.
The dimeric borazaromatic compound 32 was covalently self-assembled via a dehydration reaction of monomeric bis(borazaaromatic) ether 31 using 4 ˚A molecular sieves in acetone, as determined by 1H NMR, X-ray, and MALDI-TOF mass spectrometry (Scheme 14) (1999CC2279). The latter
method was used to monitor the dehydration reaction over 12 days. Acyclic oligomers 33 were found to predominate as the initial kinetic products
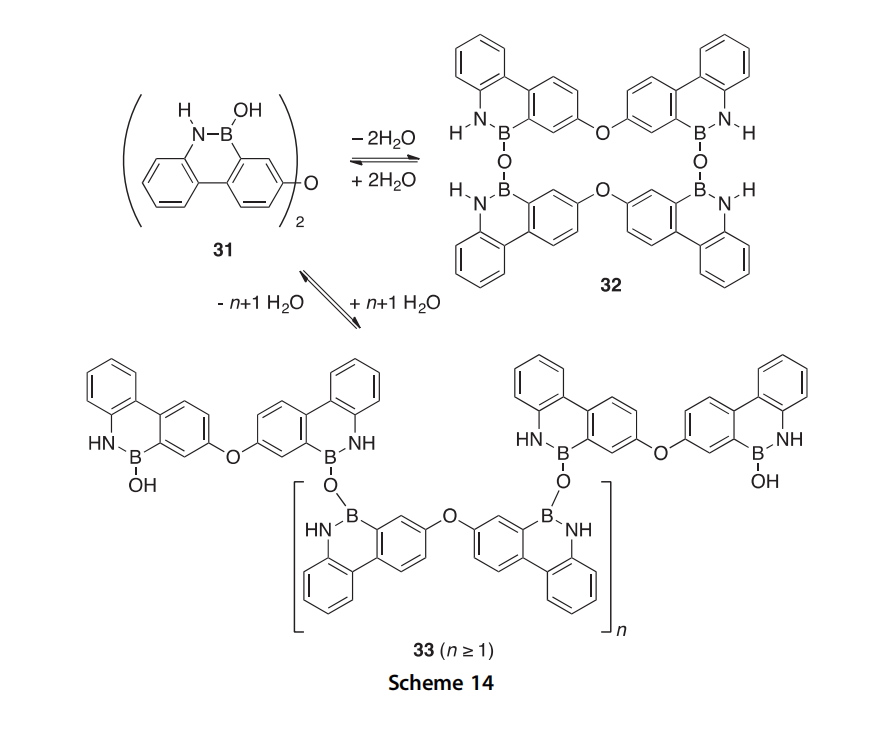
within the first 4 days, but the cyclic dimer 32 was the exclusive thermody- namic product at the experiment’s conclusion. Characterization by X-ray crystallography found that the desolvated crystals existed entirely as cyclic dimer 32, which was found to be in a helical rather than face-to-face confor- mation. Heating of the crystals for several hours at 150 ○C resulted in a mixture of the monomer, cyclic dimer, and surprisingly only oligomers 33 with an even number of monomer units in the solid state. (In solution, oligomers with an odd number of units were detected.)
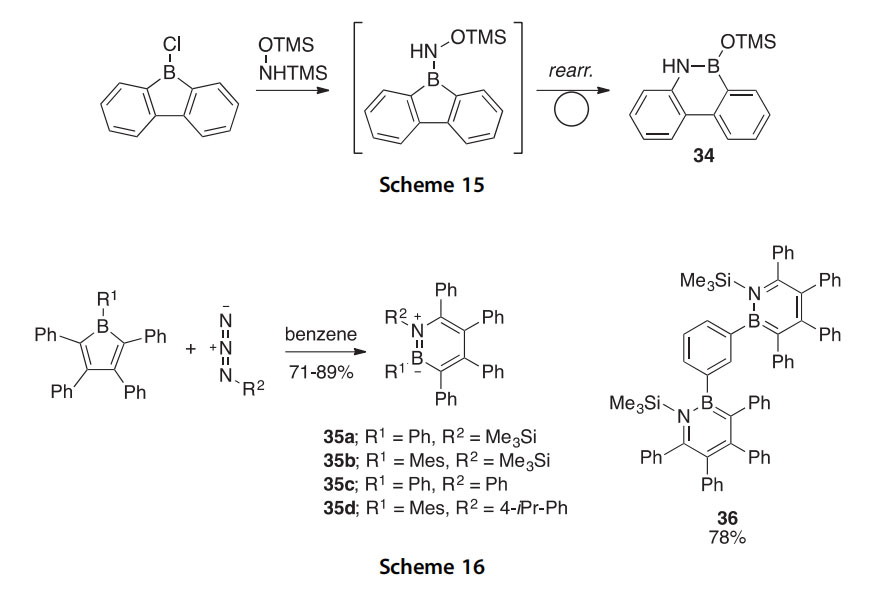
9-Chloro-9-borafluorene was treated with N,O-bis(trimethylsilyl)hy- droxylamine, producing 10-trimethylsilyloxy-9-aza-10-boraphenanthrene 34 via a room temperature rearrangement of the antiaromatic intermediate into the aromatic product 34; the reaction was monitored by NMR (Scheme 15) (2012CC4564).
This ring expansion can also be carried out by reacting boroles with appropriately functionalized azides to produce the 1,2-azaborinines 35a-d (71e89% yield) and 36 (78% yield) (Scheme 16) (2014CEJ9858).
A general method for the preparation of various monobenzo-fused 1,4-azaborines, based on the displacement of a B-methoxy group with Grignard reagents, has been reported (Scheme 17) (2014AG(I)6795). The products 37aee were isolated in 70e91% yield. When 2-pyridyllithium was used instead of the Grignards listed in Scheme 17, the product formed
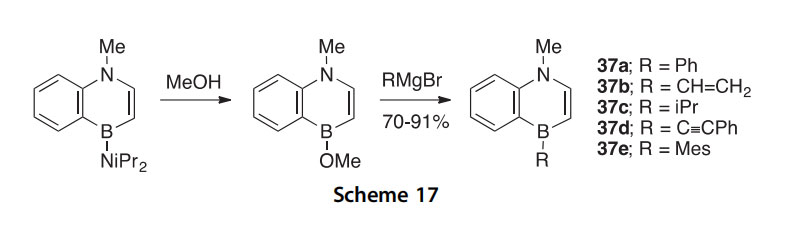
a dimer based on interaction of the B-atoms with the pyridine N-atoms. Further work targeting pyridine-based products led to the isolation of some unusual Pt-complexes that were characterized by X-ray crystallography.
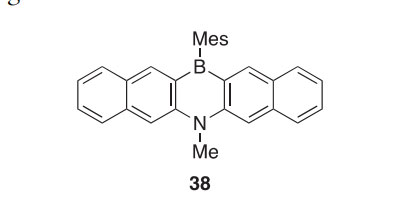
A 1,4-azaborine with two naphthalene ring fusions (38) was synthesized and shown to be stable as a radical anion during electrochemical measure- ments, highlighting its potential utility as an anion sensor and electron acceptor (2010CL612). X-ray crystallography of compound 38 revealed it to have a butterfly-like structure with a 15○ angle between the planes of the naphthalene rings.
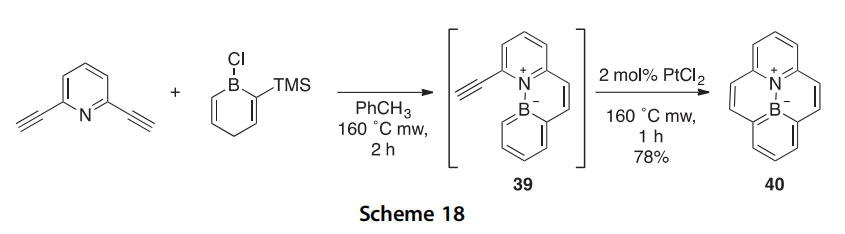
The synthesis of the BN bond for CC bond analog of the polycyclic aromatic hydrocarbon (PAH) pyrene (10b-aza-10c-borapyrene, 40) reported earlier (2007AG(I)4940) was greatly improved (from 41% to 78% yield) using microwave enhancement under carefully controlled con- ditions established by a condition-variation 1H NMR study (Scheme 18) (2014TL445). The intermediate 39, which was not isolated, formed in
2 h instead of >1 day, and ring closure of this to the pyrene analog required only 1 h instead of 4 days. As toluene is nearly transparent to microwave
energy, it is possible that the reactants themselves are the primary absorbers.
Boroxophenanthrene 41 was synthesized in 66% yield by the treatment of 2-phenylphenol with BCl3 in the presence of AlCl3, and its self-assembly capabilities were assessed (Scheme 19) (2002NJC701). In the solid state, 41
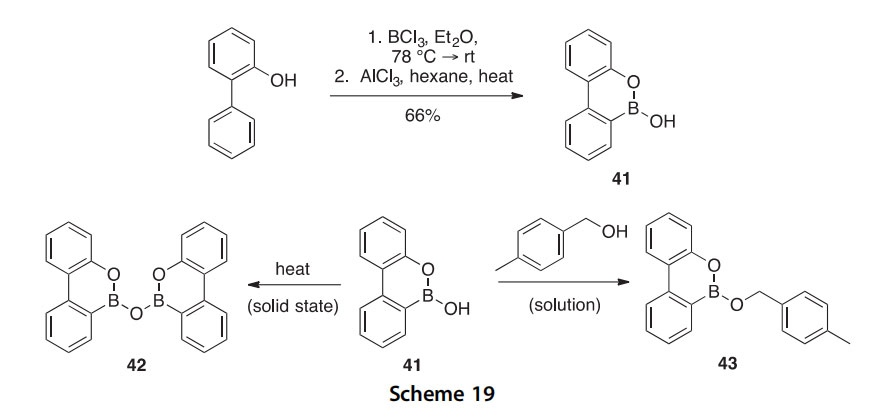
was shown to self-condense to form the homodimer 42. During 1H NMR experiments, 41 was found to react with benzylic alcohols to form alkoxy esters like 43, even at temperatures below 40 ○C, and it did not form the moisture sensitive dimer 42 under these conditions.
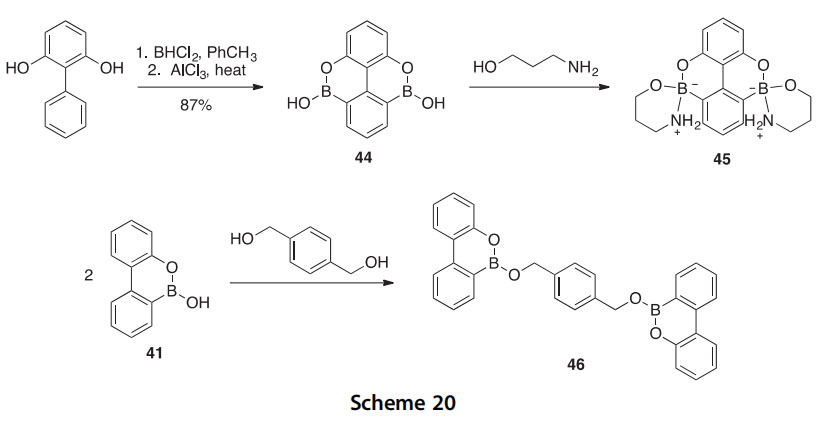
This selective reactivity led to the design of the bis-boroxoaromatic com- pound 44, which has been synthesized in 87% yield by the treatment of 2,6-dihydroxybiphenyl with freshly prepared BHCl2 followed by addition of AlCl3 and heating (Scheme 20) (2007T2391). In the solid state, 44 forms a linear polyanhydride upon dehydration. The solution-phase reactivity of compound 44 is surprisingly different from that of the monofunctional bor- oxoaromatic 41. Compound 44 proved to be unreactive with benzenedime- thanols and 1,3-propanediol, in contrast to compound 41 (see Scheme 20). The reaction of 44 with 3-amino-1-propanol resulted in the formation of compound 45 where rehybridization of the boron center from sp2 to sp3 was indicated by a corresponding upfield shift of the boron resonance in
the 11B NMR spectrum. The boron center retained its sp2 hybridization when 41 was reacted with 1,4-benzenedimethanol to form 46 (Scheme 20). The diboradiazaaromatic analog 47 of isophthalic acid was synthesized in 22% yield under similar reaction conditions used for the synthesis of 44, and it was discovered to readily undergo spontaneous dehydration to form linear oligoanhydride 48 (Scheme 21) (2001JCS(P2)2166). This reaction occurs irreversibly in the solid state after mild heating but reversibly in solution under appropriate conditions. An X-ray crystal structure of compound 47 revealed a complex network of hydrogen bonding that brought recognition sites within close proximity, thus facilitating dehydration to form linear
oligoanhydrides of MW > 3000 Da.
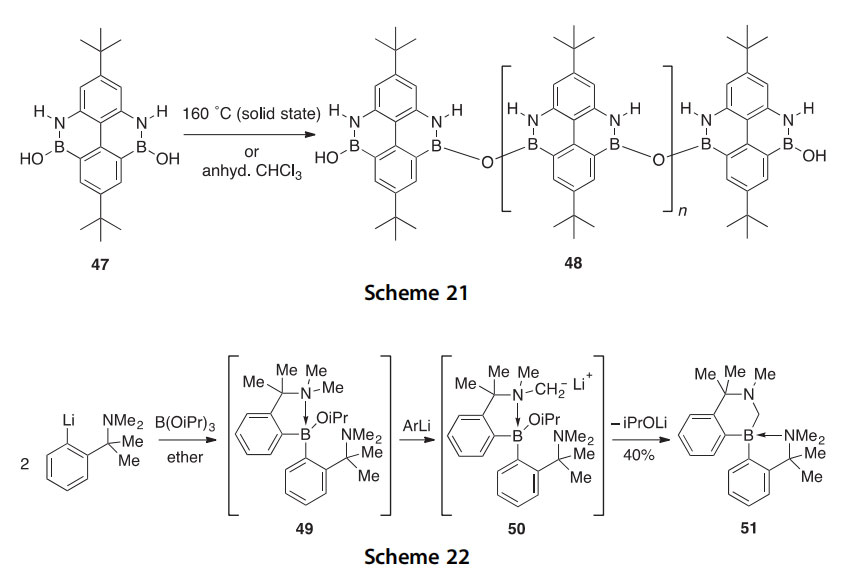
A tetrahedrally coordinated benzazaborine is generated as a major product 51 in 40% yield by the reaction of three equivalents of an aryllithium with a trialkyl borate (Scheme 22) (2000OM206). In a plausible mechanism, the treatment of B(OiPr)3 with two equivalents of the aryllithium generates diarylborane 49 which upon treatment with a third equivalent of the aryl- lithium does not generate a triarylborane, but instead gives the tetrahedrally coordinated borane species 50. This is probably due to the increased acidity of the a-protons on the tertiary amine as a result of the coordinate covalent B/N bond. Subsequent loss of an alkoxide leads to the formation of 51, and its structure has been confirmed by X-ray crystallography and 1H NMR spectroscopy.
Analogs (52) of naphthalene in which a CeC bond is replaced with an isoelectronic BN bond can be prepared by the reaction of 2-aminostyrene with dihaloboranes or alternatively by the reaction of (PhNMe)BCl2 with phenylethyne (Scheme 23) (2004ZAAC2632). Various nucleophilic substitution reactions have been carried out on the electrophilic B site of the chlorinated products including a reaction with water yielding an equi- librium mixture of the anhydro dimer 53 and the boronic acid 54. Lithiation of the t-butyl derived product 52 in the presence of N,N,N 0,N 0-tetra- methyl-1,2-ethylenediamine (TMEDA) followed by borylation with B2Cl2(NMe2)2 affords the diborane 55.
Reaction of compound 56 with Li2O yields the anhydro dimer 57, which is the N,N-dimethyl derivative of anhydro dimer 53, and this provides another route to the anhydro dimers. In analogy with the reaction to form the 1-azonia-2-boratanaphthalene 56, 1-oxonia-2-boratanaphthalenes 58aef are formed by the reaction of phenylethyne with arylchloro(phenyloxy)boranes (Scheme 23).
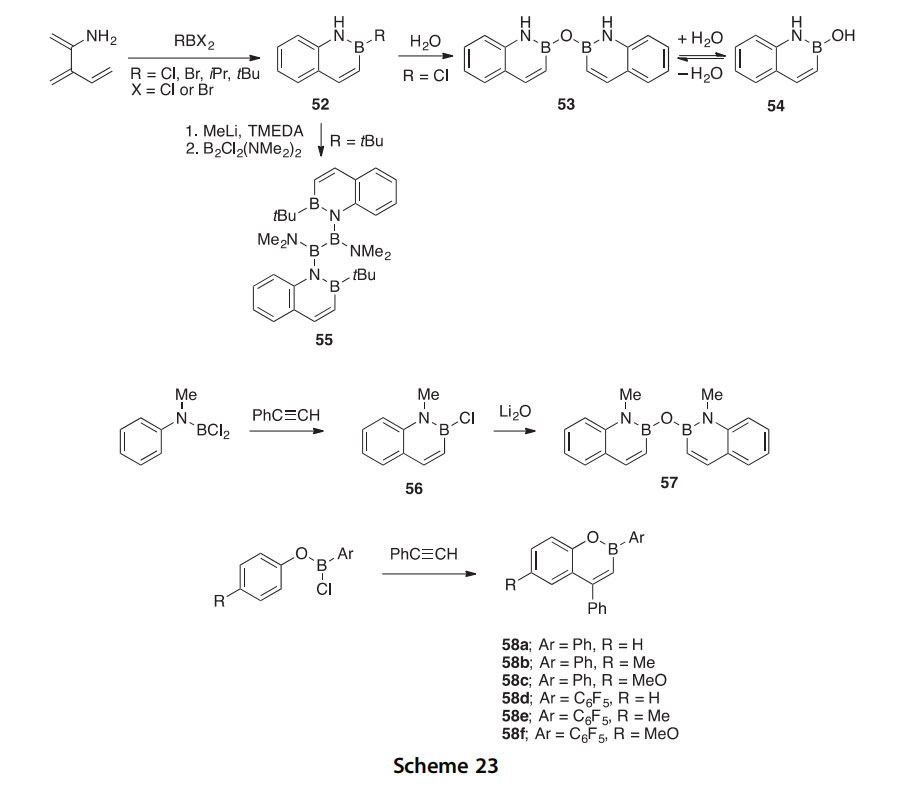
Novel p-conjugated ladder-type fused azaborines (59e61) have been synthesized in 46e54% yield, first by generation of a tetralithio species followed by reaction with MesB(OMe)2 at reflux (2006OL2241). These compounds were characterized by UVevis and fluorescence spectroscopy. The so-called “para-type” compounds 60 and 61 exhibited red-shifted absorption maxima compared to reference compound 62 due to extended p-conjugation while “meta-type” compound 59 exhibited a comparable absorbance to 62 indicative of weak to no p-conjugation between adjacent azaborine units. The “para-type” compounds are potentially useful as organic light-emitting devices due to their high quantum yields upon photoexcita- tion. X-ray crystallographic data of structure 59 revealed that this compound exists as a nearly planar molecule and surprisingly did not show any intermo- lecular interactions like pep stacking or CHep interactions. It is speculated that perhaps the bulky mesityl groups attached to boron are responsible for discouraging such interactions. Work was also done to introduce different functional groups onto the periphery of these compounds (2007CC3204). In follow-up studies, thiaborin units (in which S replaced N in azaborines 59e61) were also synthesized and these, along with azaborines 59e61, were tested for their fluoride and/or cyanide anion sensing ability (2007CEJ8051, 2009JOM3833, 2009CC1894, 2009CEJ5056). The Lewis
acidity of the boron centers can be attenuated by extension of the p system or by exchange of nitrogen for sulfur. Additionally, the multistep formation of complexes between these compounds and anions can be monitored by UVe vis and fluorescence spectrophotometry (sometimes by visual color changes in fluorescence emissions), making azaborines potential candidates for anion detection in both biological and industrial settings.
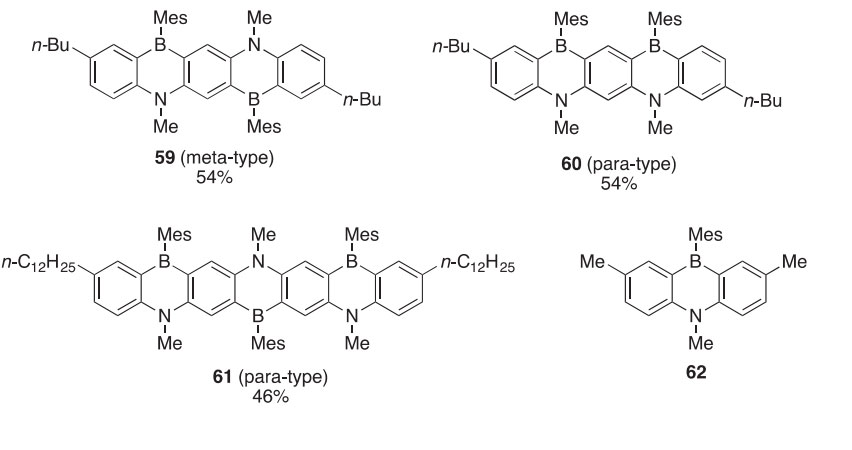
p-Conjugated dendrons (63e65) and dendrimers (66) based on diben- zoazaborines were synthesized, providing the first donoreacceptor-type compounds of this type (2009OL3534). The branching and terminal precur- sors (67 and 68, respectively) were synthesized using a series of halogen metal exchanges and coupling reactions. Dendrons 63e65 were synthesized via Pd-catalyzed coupling reactions between 67 and 68. The dendrimers (66) were obtained in 13e62% yields by basic alcoholysis of the trimethylsilyl (TMS)-protected dendrons followed by Pd-catalyzed coupling. The den- drons exhibited strong absorption and photoluminescence properties, which indicate that the azaborine units are positioned perpendicularly to each other. The dendrimers exhibited varying strengths of fluorescence emissions due to photoinduced electron transfer from the azaborine dendrons to the benzo- thiadiazole core unit, a so-called intramolecular charge-transfer state.
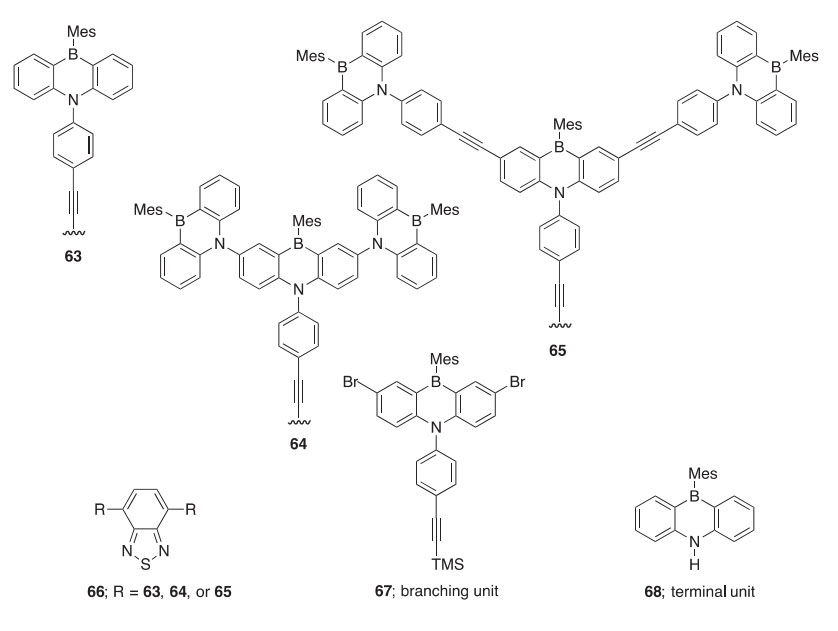
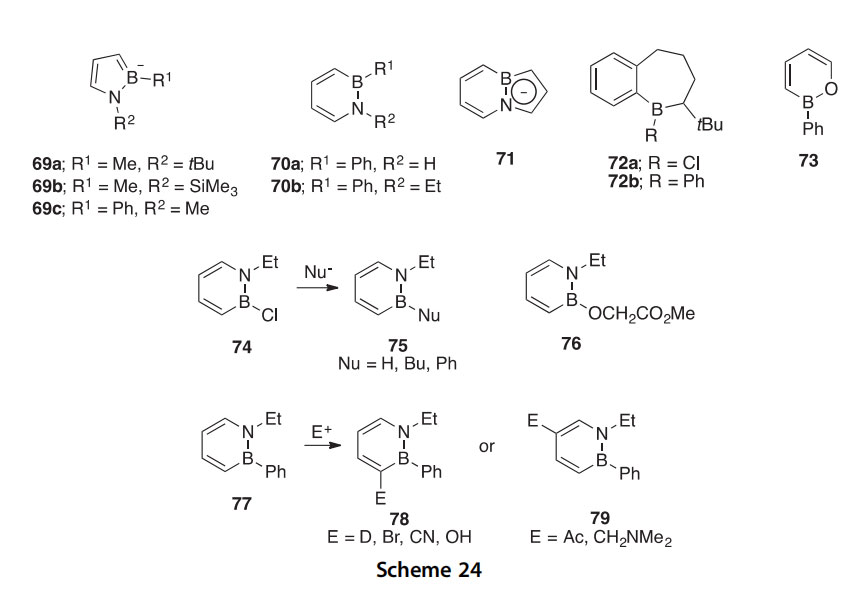
Heteroaromatic 1,2-azaborolides 69aeb and 1,2-azaborines 70a,b have been synthesized via RCM followed by oxidation by 2,3-dichloro-5,6- dicyano-1,4-benzoquinone and were confirmed to be weakly aromatic by 1H, 11B, and 13C NMR (2000OL2089). A heteroaromatic analog of indene (71), in which the ring junction C]C bond is replaced by a BN bond, has been synthesized by similar procedures followed by deprotonation using KN(SiMe3)2 (2002OM4578). This is converted to azaboronaphthalene upon ring expansion with n-BuLi/CH2Cl2 (2006OM513). The monocyclic 2-(diisopropylamino)-2H-1,2-thiaborinine has been synthesized in similar fashion using lithium diisopropylamide (LDA)/CH2Cl2 (2011OM3698). Alternatively, 1,2-azaborolides can be synthesized after Sn/B exchange using a commercially available azastannole (2004OM5626). This Sn/B exchange re- action has also been applied to the synthesis of borepines 72a,b (2000JCS(P1) 1965). 1,2-Azaborines can be synthesized via a carbenoid ring expansion of the 1,2-azaborolide with LDA/CH2Cl2 (2001OM5413). Oxaborine 73 was also synthesized via Sn/B exchange followed by carbenoid ring expansion (2007OM1563). The azaborolides, azaborines, the azaborindenyl, and the oxaborine form complexes with heavy metals similar to their respective parent compounds, which is further indication of their p-delocalized structures. A BN isostere of benzene (74) has also been synthesized via an RCM reaction followed by oxidation (2007OL4905). Nucleophilic substitutions have been carried out at the electrophilic B-atom of 74, providing a method for the syn- thesis of a wide range of B-substituted 1,2-azaborines (75), including an iso- stere of a hypolipidemic agent (76) (Scheme 24). The 1,2-azaborine 77 gives the three- or five-substituted derivatives 78 and 79, respectively, via electrophilic aromatic substitution (EAS) reactions (2007OL679).
In a study of the reactivity of 1,2-dihydro-1,2-azaborine 80, it was found
that this BN for C]C replacement mimic of benzene undergoes SNAr
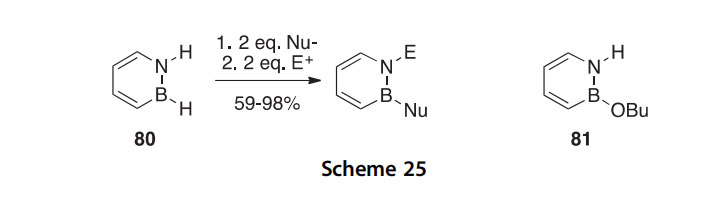
reactions (unlike benzene) giving products (59e98% isolated) in which the nucleophile (nBu, tBu, Ph, vinyl, OtBu, O-allyl) is attached boron and the electrophile (TMS, H, Me) is attached to nitrogen (Scheme 25) (2011AG(I)8157). The protecting group-free synthesis of the versatile 1,2- azaborine synthon 81, useful for the preparation of BN-benzenoids, was reported in a separate publication (2013JA12908).
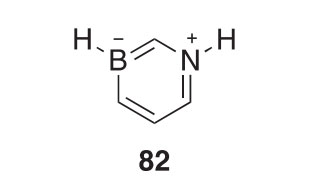
In a related study, 1,3-dihydro-1,3-azaborine 82 was synthesized and proved to be a stable structural motif with considerable aromatic character (2011JA2152). By X-ray crystallography, 82 was shown to have significant electron delocalization. Nucleophilic substitution occurs at the B-atom and EAS reactions occur at the C-atom across the ring from the boron.
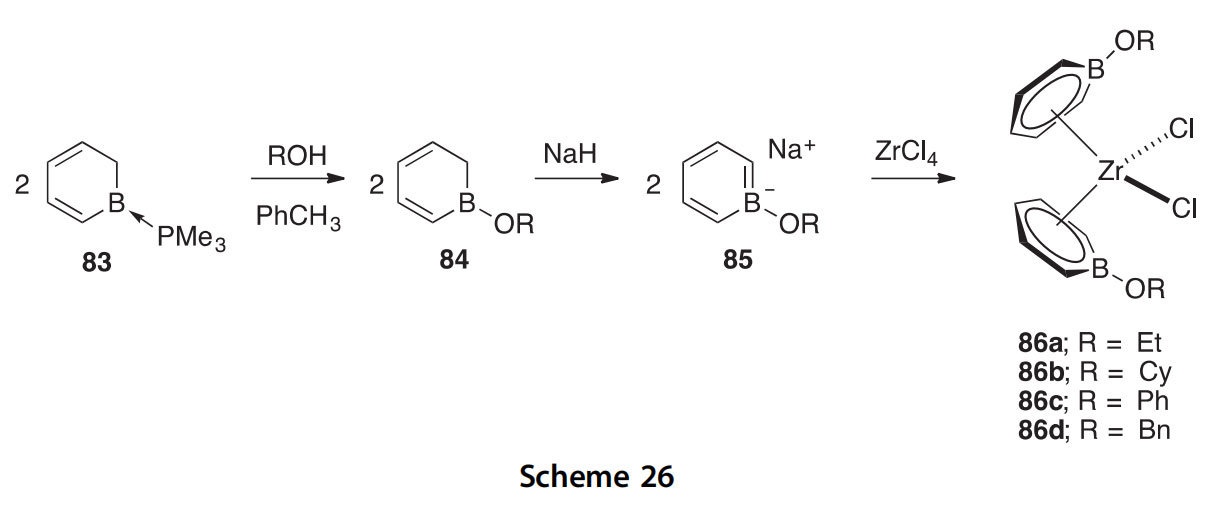
The trimethylphosphine adduct 83 of borabenzene was treated with a series of alcohols to give boracyclohexadienes 84, which upon deprotona- tion give the alkoxyboratabenzene ligands 85; coordination with zirconium produces the metallocenes 86aed (Scheme 26) (1999JA1288). These com- pounds are of interest for their ability to react with excess methylaluminox- ane under 1 atm of ethylene to give alkene mixtures. The structure of metallocene 86c was determined crystallographically.
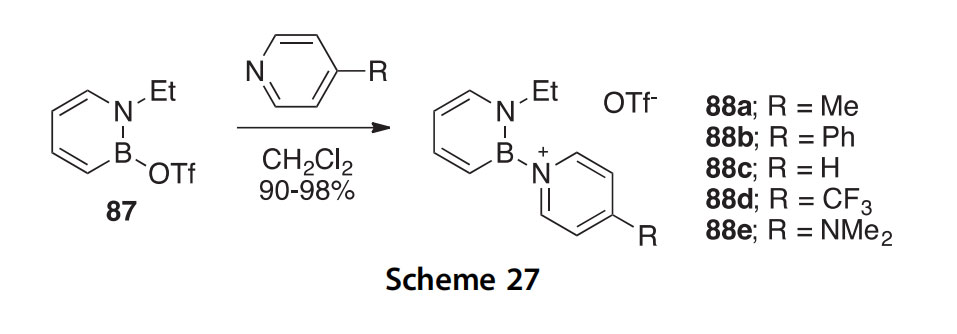
A set of highly fluorescent boron heterocycles 88aee have been synthe- sized in 90e98% yield from the triflate 87 (Scheme 27) (2010AG(I)7444). Absorbance lmax values are 283e292 nm, while emission values are 436e537 nm. The X-ray crystal structure of 88b was determined.
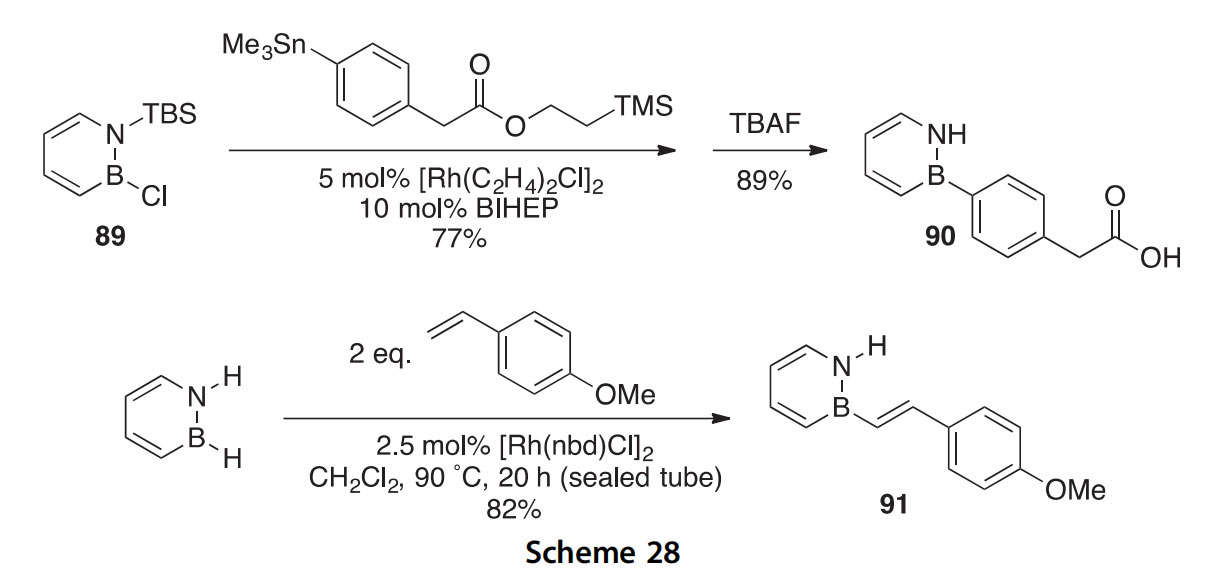
A method for the B-arylation of boron heterocycle 89 has been devel- oped and used to synthesize the BN for C]C analog 90 of the nonsteroidal anti-inflammatory drug (NSAID) felbinac (Scheme 28) (2013AG(I)9316). An X-ray crystal structure of the immediate precursor to the acid 90 was determined. The synthesis of the B-N for C]C analog 91 of 4-methoxy-trans-stilbene has been accomplished in a similar Rh-catalyzed dehydrogenative coupling reaction (2014OL3340). An X-ray crystal struc- ture of the N-benzylated derivative of product 91 was reported.
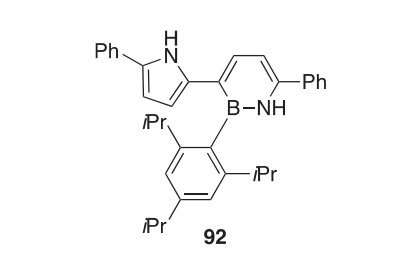
The azaborine derivative 92 was synthesized unexpectedly by the treat- ment of N-Boc-protected bis(5-phenyl-2-pyrrolyl)borane with BF3$Et2O (2010OM5732). A careful analysis of the B-N bond lengths elucidated by X-ray and density functional theory (DFT) calculations revealed that the aza- borine ring behaves more like a cyclohexadiene analog than a benzene analog. The absorbance (lmax 404 nm) and emission (lmax 481 nm) spectra give results that are demonstrative of an extended p-conjugated system.
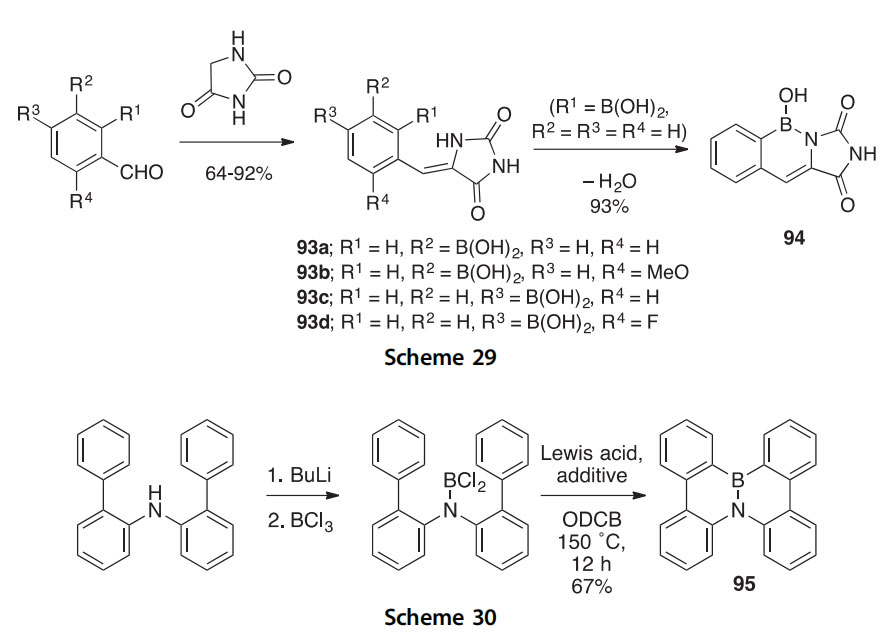
The unexpected fused azaborine 94 was obtained in 93% yield via an aldol condensation/dehydration reaction of hydantoin with 2-formylphenylboronic acid (Scheme 29) (2010HCA1093). This discovery was made while examining the condensation products (93aed) of hydantoin with other formylphenylboronic acids in a search for new biologically active boron-containing compounds. The structure of 94 has been confirmed by X-ray crystallography. Attempts to synthesize a similar fused azaborine starting with 3-formyl-2-thiopheneboronic acid gave only a deboronated product.
In a similar effort, the BN for C]C isosteric replacement mimic 95 of dibenzo[g,p]chrysene was prepared in several steps from bis(biphenyl-2-yl) amine and its X-ray crystal structure determined for a direct comparison with that of the parent PAHs (Scheme 30) (2011JA18614). A condition- variation study of the last step revealed that 4 equivalents of Lewis acid AlCl3 and 1.5 equivalents of the additive 2,2,6,6-tetramethylpiperidine
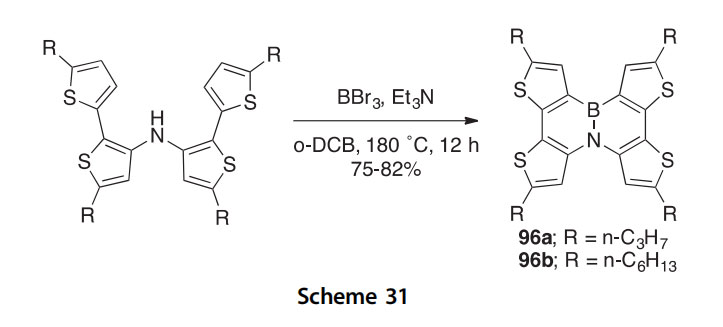
give the best yield of 95 (67% isolated). A double BN for CC isosteric replacement mimic of hexabenzo[a,c,fg,j,l,op]tetracene was also prepared. The BN-substituted PAHs may prove to be useful in the preparation of electronic materials such as semiconductors.
A further advance in this field was made when the two tetrathieno-fused naphthalene BN for C]C analogs 96a,b were synthesized and investigated (Scheme 31) (2013AG(I)3117). Boron heterocycle 96a was studied by X-ray crystallography and DFT calculations, both of which showed evidence of a high degree of aromaticity explaining the observed chemical stability. The first organic electronic devices based on these azaborine com- pounds were found to have a high field-effect mobility of 0.15 cm2/V s, thus highlighting the great potential of azaborine chemistry to produce novel organic semiconductors.
The azaborines 97aec and 98 have been synthesized from nonaromatic heterocyclic precursors via photoeliminations resulting from unusual and unprecedented breakage of CeH and BeC bonds (Scheme 32)
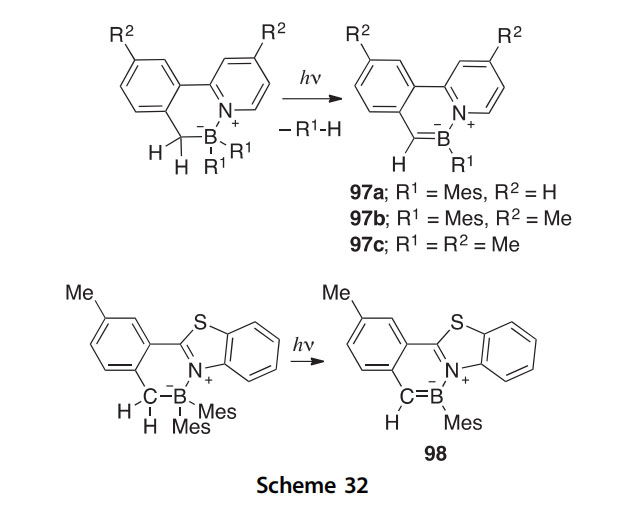
(2013AG(I)4544). This reaction has even been shown to occur in the solid state when polymer films such as poly(methylmethacrylate) or poly(N- vinylcarbazole) are doped with the precursors, potentially providing a convenient method for the development of materials with optoelectronic applications.
3. DIHETERABOROLES AND DIHETERABORINES
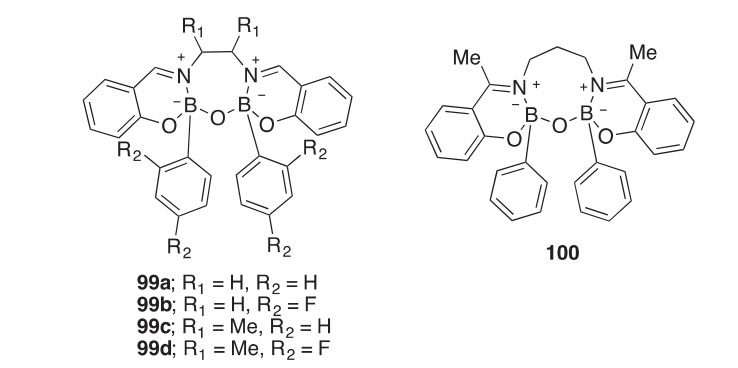
Oxazaborepines 99aed and oxazaborocine 100 have been synthe- sized in 38e95% yields by the cyclocondensation of a boronic acid with different salen ligands (2001IC6405). The open forms of macrocycles synthesized from salen, acen, or acpen ligands can be obtained directly by carrying out the reactions in refluxing EtOH or through alcoholysis of the macrocycle. Salphen, salcen, and salpen ligands only generate the macrocyclic compounds. X-ray crystallographic analysis shows that the oxazaborepines favor an undistorted chair conformation and the oxazabor- ocines favor a boat conformation.
A synthesis of 6-aryl-8H-dibenzo[d,h][1,3,7,2]dioxazaborecin-8-ones, which are bridgehead bicyclo[4.4.0]boron heterocycles of general structure 101 (R1 H, Cl; R2 H, F, Et; R3 2-OMe, 4-Br), has been developed and features a one-pot three-component reaction of a 2-aminobenzoic acid, a 2-hydroxybenzaldehyde, and an arylboronic acid (Scheme 33) (2012T3377). The reaction is conducted in CCl4 under microwave irradi- ation and gives the bridgehead bicyclo[4.4.0]boron heterocycles 101 in 95e98% yields. One of the compounds (101, R1 R2 R3 H) was solved crystallographically. A similar three-component additive-free method
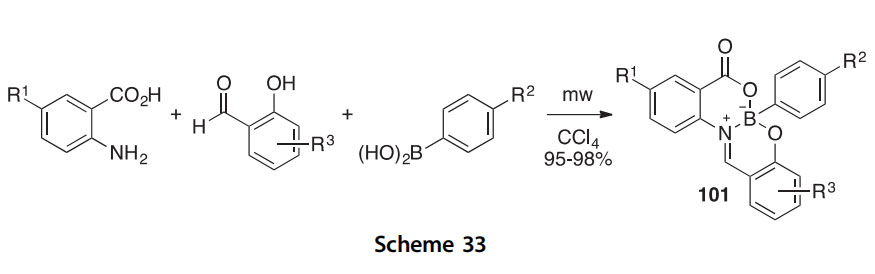
has been developed for condensing boronic acids with phenols and alde- hydes under solvent-free conditions (2010AJC6519).
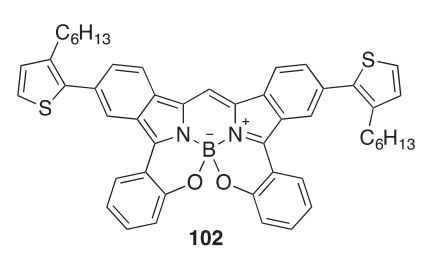
Thiophene-conjugated BODIPY dye molecules with benzo[1,3,2] oxazaborinine rings, such as 102, have been synthesized and found to absorb near-infrared (NIR) light with relatively high molecular extinction coefficients (2011OL4574). These can serve as light-harvesting sensitizers in polymeric solar cells.
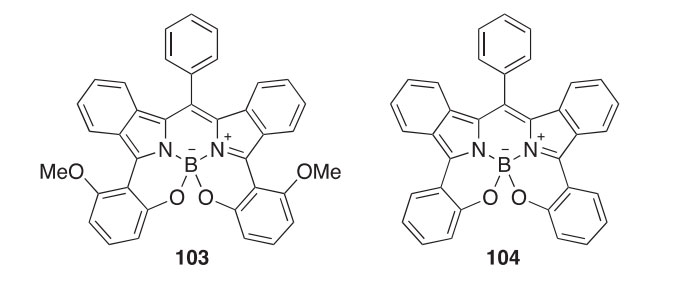
In a related study, several O-chelated meso-phenylboron-di(2H- isoindolyl)methenes, e.g., 103 and 104, were synthesized and found to have NIR absorption and emission (600e850 nm) (2011T3187). X-ray crystal structures of both compounds were presented. The latter has a fluo- rescence lmax emission at 733 nm and a F quantum yield value of 0.58.
The boron heterocycles 106e108 can be synthesized by the reaction of the eight-membered rings 105 with n-BuLi and an alkyl halide giving a mixture of products in which (1) the alkyl group at C2 can be either exo
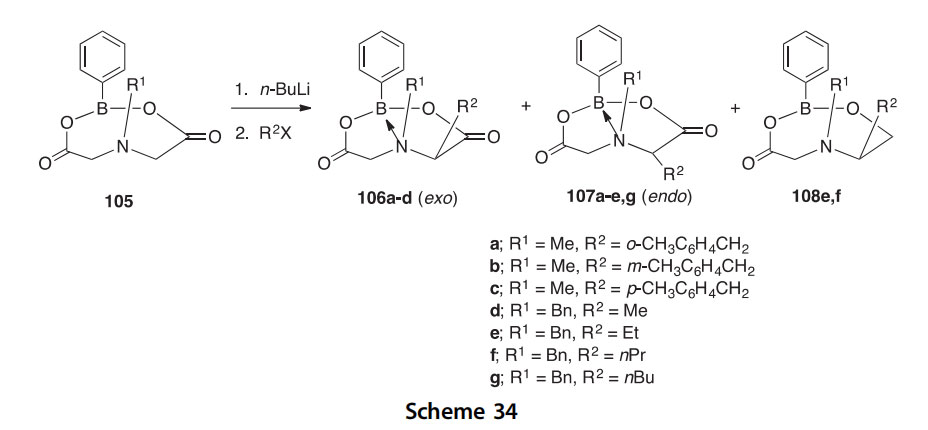
or endo (106 and 107), or (2) there is no N/B cross-ring chelation (108) (Scheme 34) (2007POL1023).
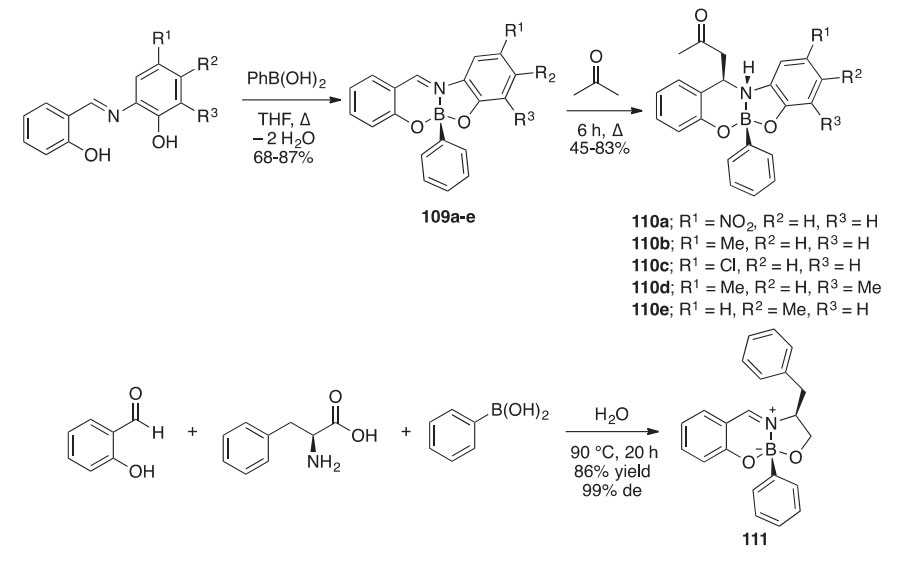
Diazaborocines 110aee were synthesized stereoselectively in 45e83% yield via an acetolysis reaction at the C]N of the Schiff base derived [4.3.0] boron heterobicycles 109 (Scheme 35) (2001CJC1229). The reac- tion of N-salicylidene-2-aminophenolate (SAP-H2) derivatives with phe- nylboronic acid afforded the five [4.3.0] boron heterobicycles 109aee in 68e87% yields. Dative bonding between the nitrogen atom of the Schiff base and boron atom on compounds 109aee increased the polarity of the C]N bond, thus facilitating the aldol condensation of acetone and the boron heterobicycle to afford diazaborocines 110aee. X-ray crystallog- raphy of derivative 110d revealed that the compound exists as a dimer stabilized by hydrogen bonding. Similar heterocyclic Schiff bases were unintentionally prepared during an exploration of the viability of the Petasis-borono-Mannich multicomponent reaction in water (2010T2736). The reaction of L-phenylalanine and salicylaldehyde with phenylboronic acid in water for 20 h at 90 ○C afforded 111 in 86% yield with a 99% de. This approach has been extended to the synthesis in 31e96% yield of the similar derivatives 112aeg by combining L-phenylalanine with different salicylaldehydes and boronic acids.
Stable boron heterocycles sometimes arise unexpectedly. The imine 113, produced from salicylaldehyde and NH4OAc in the presence of dimethyl malonate, undergoes reduction with NaBH4 or NaBH3CN or NaBH(OAc)3 to give the cyclic boramide 114 (Scheme 36) (2014T8614). The X-ray crystal structure of this fairly stable compound has been determined; mechanistic studies revealed an oxazaborinane intermediate in the reaction pathway.

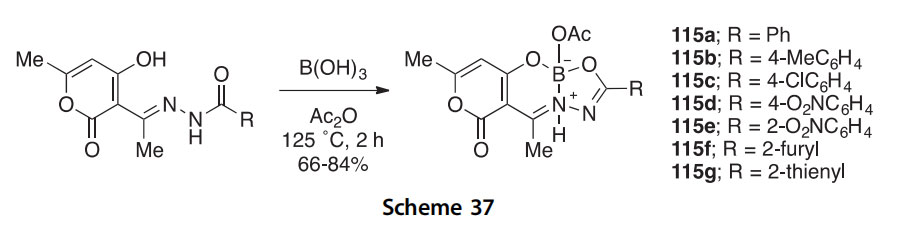
A similar investigation looked at the condensation of dehydroacetic acid N-aroylhydrazones with B(OH)3/Ac2O mixtures (known to produce B(OAc)3) to give the boron heterocycles 115aeg in 66e84% yield (Scheme 37) (2015T7245). An X-ray crystal structure determination of derivative 115a was obtained.
The oxazaborolo-benzoxazaborininone derivatives 116 of resorcinar- ene were synthesized in 50e75% yields and in 98% de via a Mannich reaction with L-proline followed by treatment with a boronic acid or ester (Scheme 38) (2003TA2787). Introduction of the electrophilic boron atom into this structure opens up a potential use for the catalysis of asym- metric reactions. The derivatives 116 were determined to exist as either crown or diamond conformers according to 1D and 2D 1H NMR exper- iments. Similar derivatives can also be prepared from (1S,2R)-ephedrine (2004MI75).
The reactions of 9-Cl-9-borabicyclo[3.3.1]nonane (9-Cl-9-BBN) or 9-MeO-9-BBN with MeSCH2Li$(TMEDA)n have been investigated, leading to the isolation of the unprecedented boronesulfur heterocycles 117 and 118 (Scheme 39) (2014OM4336). The former product 117, an inner salt formally composed of sulfonium and borate subunits, rearranges
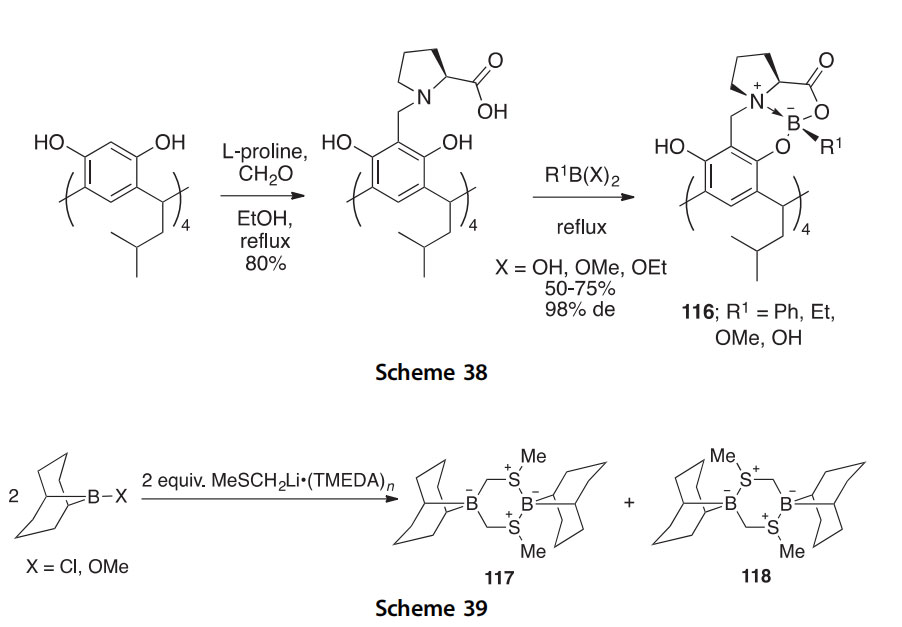
upon heating to the thermodynamically more stable Ci-symmetric 118. Both of these unusual boron heterocycles have been studied by 1D and 2D 11B NMR as well as X-ray crystallography.
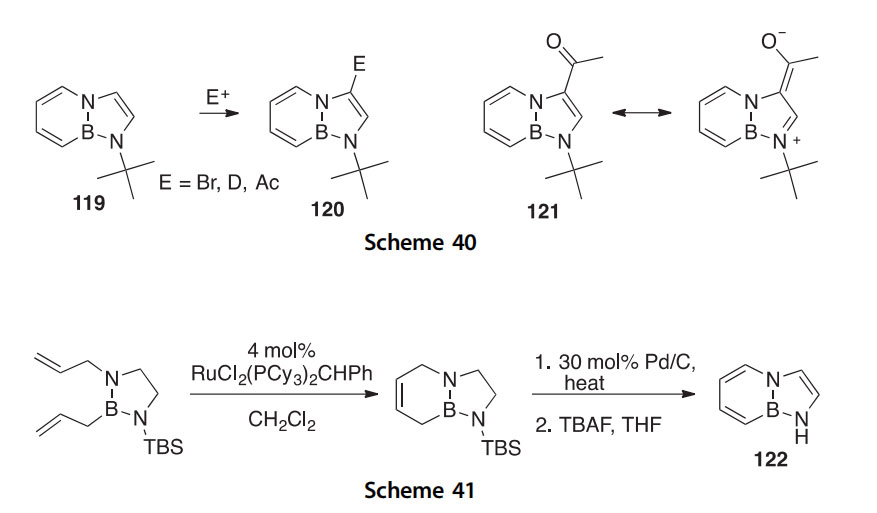
A BN for C]C replacement analog (119) of an N-protected indole was synthesized via an RCM reaction followed by a dehydrogenation in which it was the ring fusion bond that was replaced (Scheme 40) (2010JA16340). EAS reactions occur faster than for the isoconjugate indole compounds but with the same regioselectivity (i.e., to give 120). As part of the characteriza- tion, X-ray crystallography of several products was conducted, including that of the 3-acetylated compound 121, which proved to have a structure consistent with a significant contribution by its zwitterionic resonance hybrid (Scheme 40).
Synthesis of the parent “fused” BN indole 122 was accomplished using an RCM route (Scheme 41). This BN for C]C isosteric replacement mimic of indole was thoroughly compared to indole itself using structural (X-ray), acidity (pKa), electronic (cyclic voltammetry), and spectrophoto- metric (UV, fluorescence) methods (Scheme 41) (2011JA11508).
The diazaborines 123aec have been synthesized in 55e78% yields by reacting aniline derivatives with AlCl3 followed by the addition of BCl3 with heat (Scheme 42) (2002TL3255). By monitoring the reaction progress through HPLC, it was found that an amidine is generated in situ after the reaction between the AlCl3 and aniline. This facilitates ring closure by EAS upon addition of gaseous BCl3 allowing for the formation of the dia- zaborines. X-ray crystallographic analysis confirmed the structure of
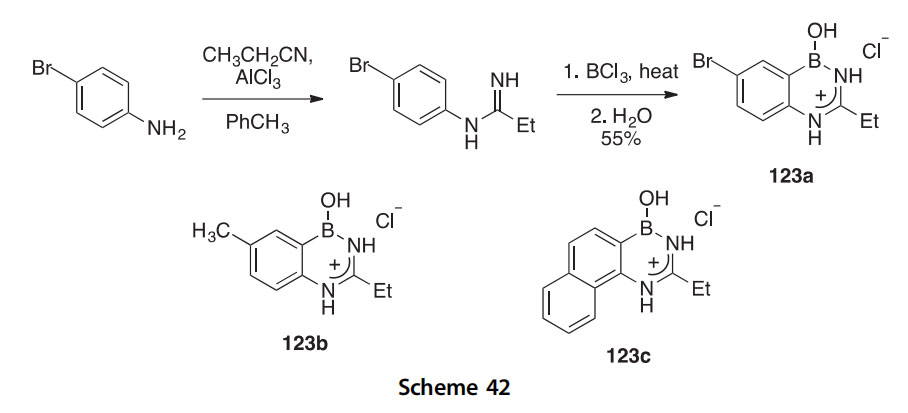
derivative 123a as a hydrochloride salt. This synthesis is also applicable to p-toluidine and a-naphthylamine to afford 123b and 123c.
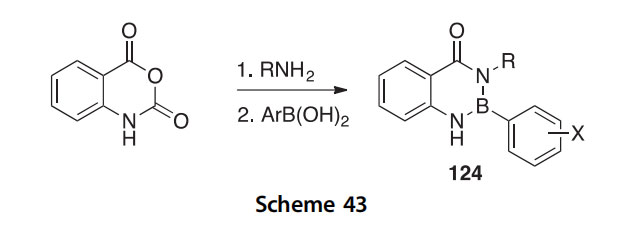
A set of benzo[d][1,3,2]diazaborinin-4(1H)-ones 124, boron-containing quinazolinones, were prepared in a solvent-free, two-step, one-pot reaction starting with the condensation of isatoic anhydride and amines to give 2-aminobenzamides, followed by reaction with boronic acids (Scheme 43) (2013SC2936).
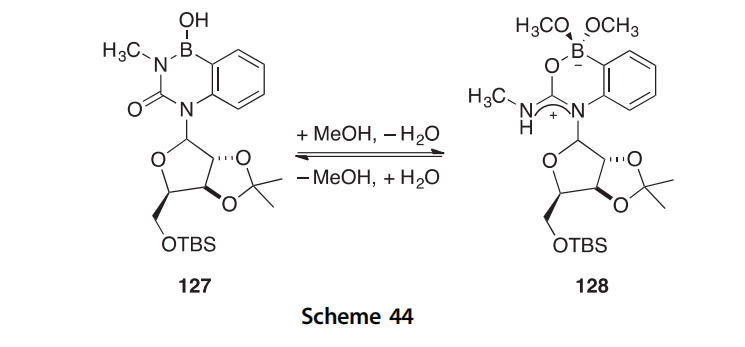
The benzo-fused 4-borauracils 125aec can be prepared in 73e87% yield by the cyclocondensation of 2-aminophenylboronic acid with isocy- anate derivatives (Scheme 44) (1999JOC9566). The benzoborauracils exist as bis-methanol adducts (126aec) in methanolic solutions according to 1H and 11B spectral analysis. The degree to which 125aec exist in equilibrium with 126aec is dependent upon the concentration of methanol and the nature of N-2 substituent. It can be inferred that the substituents that contributed to larger ring strain favored formation of the bis-methanol adducts. The structures of compounds 125b, 125c, and 126c were confirmed by X-ray crystallography. The novel benzoborauracil nucleoside 127 has been prepared, and a similar bis-methanol adduct 128 is formed under similar conditions.
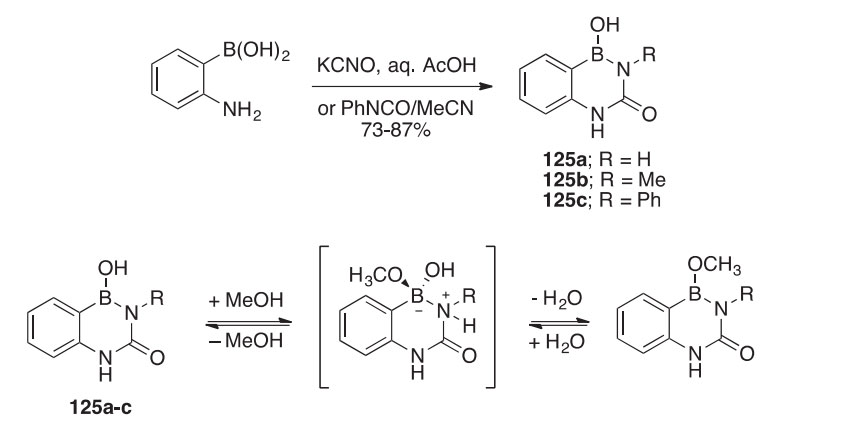
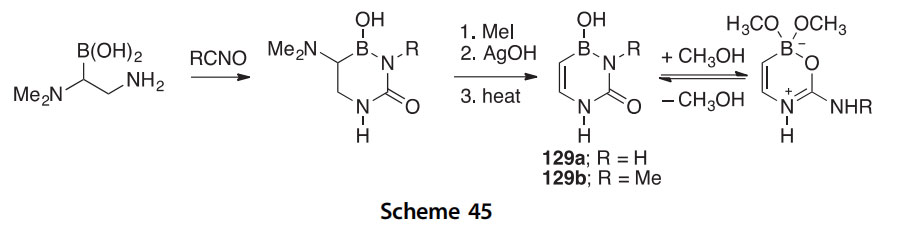
The 4-borauracils 129a,b were prepared by condensing a b-boronic amino acid with isocyanic acid or methyl isocyanate followed by Hoffman elimination (Scheme 45) (2010MI33). The 11B NMR spectrum indicated that the corresponding bis-methoxy adducts form when these compounds are treated with a large excess of methanol.
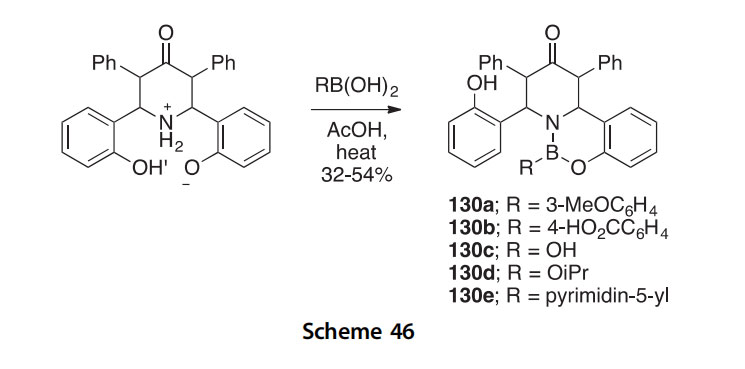
Benzopyridooxazaborinines 130aee have been synthesized in 32e54% yields by the cyclocondensation of a piperidone derivative with boronic acids in refluxing AcOH (Scheme 46) (2008CHC1527).
The solvent-free and demonstrably microwave-facilitated condensation of 1,2-diaminobenzene and 1,8-diaminonaphthalene with arylboronic acids is a rapid way to obtain the 1,3,2-benzodiazaboroles 131aef and the naphtho[1,8-de][1,3,2]diazaborinines 132aef, respectively (Scheme 47) (2013JOM122). Extensive characterization by 15N NMR (12 compounds) and X-ray crystallography (4 compounds) has provided evidence of exten- sive N/B electron delocalization in both types of boron heterocycles.
Bis- and tris-1,3,2-diazaborole derivatives 133aed and bis- and tris-1,3,2- diazaborolidines 134aee with either benzene or biphenyl cores have been synthesized and found to have interesting structural and spectrophotometric characteristics (2006JCS(D)2127). The biphenyl derivatives 133d and 134e show more pronounced optical behavior compared to that of the benzene derivatives by exhibiting a strong blue luminescence (labs ¼ 278 nm, lem ¼ 324 nm in hexane) with the exception of 133c, which exhibited a moderate luminescence at (labs ¼ 287 nm, lem ¼ 381 nm in hexane and
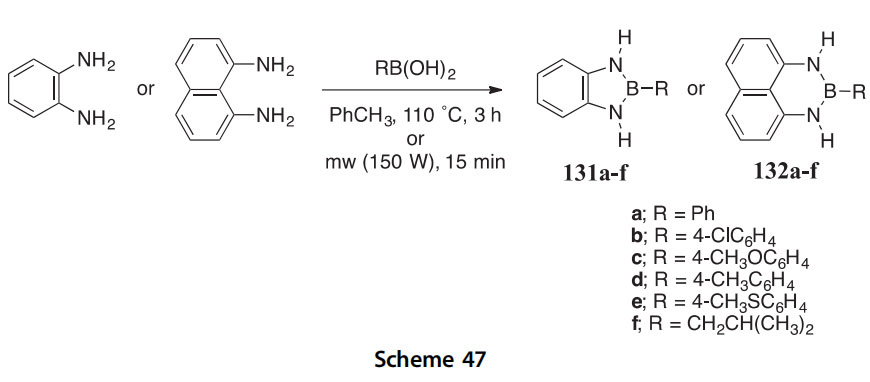
THF). X-ray crystal analyses of 133c, 134a, and 134d show that the hetero- cycles are not coplanar with the arene core, thus significantly hindering p-delocalization within these structures.
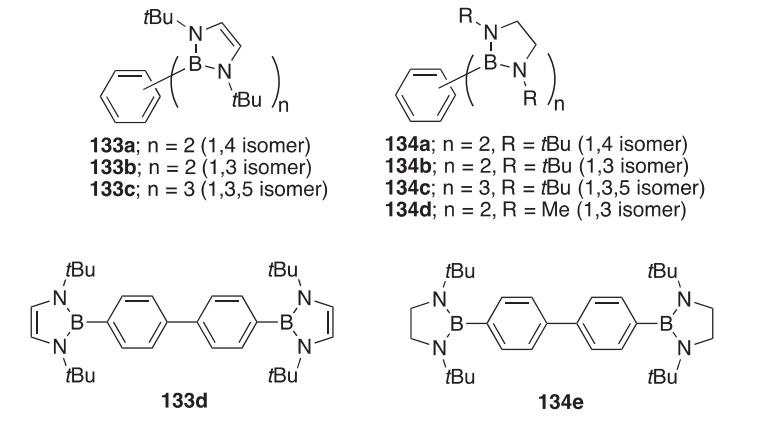
2-Formylphenylboronic acids typically condense with hydrazines to produce 2,3,1-benzodiazaborines, but with the very electron-withdrawn 2,4-dinitrophenylhydrazine in EtOH, they give instead the weakly intramo- lecularly chelated (E)-hydrazones 135a,b (Scheme 48) (2002CCC1084). Both structures were determined crystallographically. The chelated diethyl- borate 135b is hydrolyzed to the boronic acid 136 by heating in wet DMF, but this product did not dehydrate to form a boron heterocycle. On the other hand, with mono-nitrophenylhydrazines, the 4-methoxy-2- formylphenylboronic acid condenses directly to give nitrophenyl- substituted 2,3,1-benzodiazaborines 137a,b. In yet another outcome, phenylhydrazine itself first affords the intramolecularly hydrogen-bonded
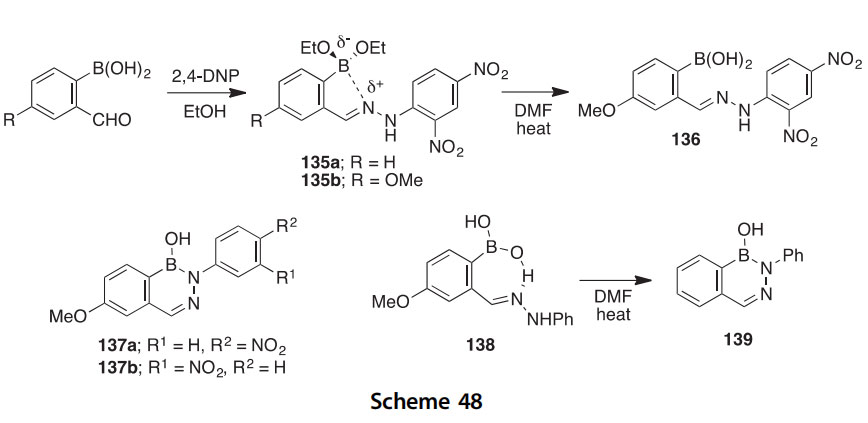
product 138, which can be dehydrated to the diazaborine 139 by heating in wet DMF (Scheme 48).
An investigation of the cyclodehydration of 2-acylphenylboronic acids with hydrazides led to the discovery of new 2-acyl-2,3,1-diazaborine het- erocycles with unusual hydration/dehydration reactivities and antibacterial properties (Scheme 49) (2014CBD1381). The structures obtained are highly dependent upon the conditions of synthesis. Open hydrazone structures like 140 are often obtained at 23 ○C with EtOH as solvent; monomer boron heterocycles like 141 sometimes form directly, and anhydro dimers like 142 are often obtained at 82 ○C with CH3CN as solvent and azeotropic removal of H2O. The crystal structure of the anhydro dimer 143, formed from 2-formylphenylboronic acid and nicotinic acid hydrazide, clearly shows its unusual double intramolecular chelate structure. Another new boron heterocycle to come out of this investigation is the pentacyclic com- pound 144, synthesized by a 2:1 condensation of 2-formylphenylboronic acid and carbohydrazide. The crystal structure of compound 144 shows a slight curvature from planarity of the five fused rings, presumably due to packing forces, and the C]O group is rather ketonelike in character by some analytical measures. Yet a third new boron heterocycle to come out of this work is the intramolecularly chelated compound 145 which is
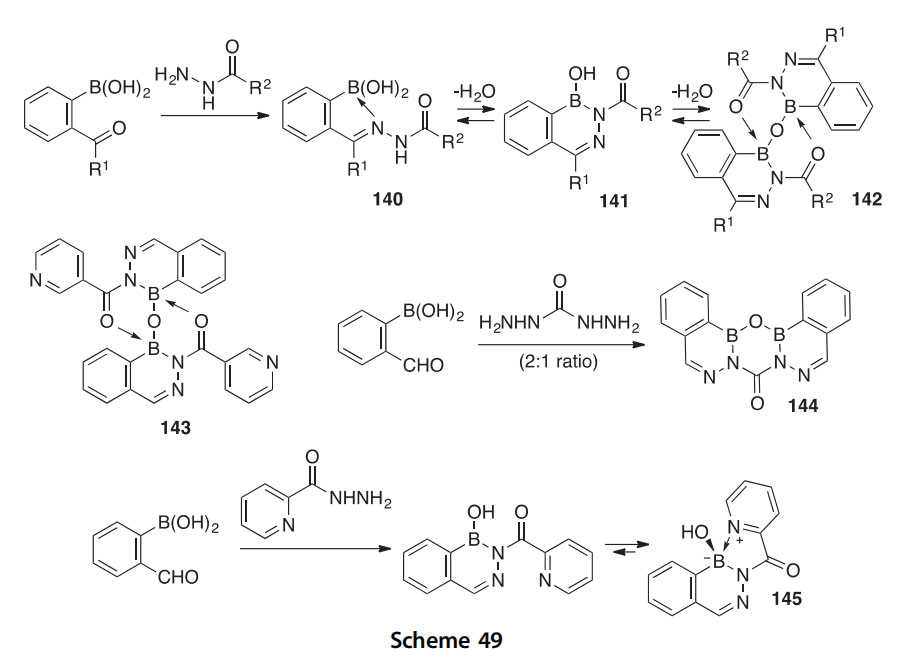
produced from 2-formylphenylboronic acid and picolinic acid hydrazide. Its structure was solved by X-ray crystallography. Heterocycle 145 undergoes an unusually slow deuterium exchange of its OH group, determined by 1H NMR, indicating that the exchange is occurring not in the chelated structure but rather in the minor (unchelated) open structure that is in equi- librium. Of the forty four compounds prepared, nine were found to possess activity against Escherichia coli, and two others were active against Mycobacte- rium smegmatis.
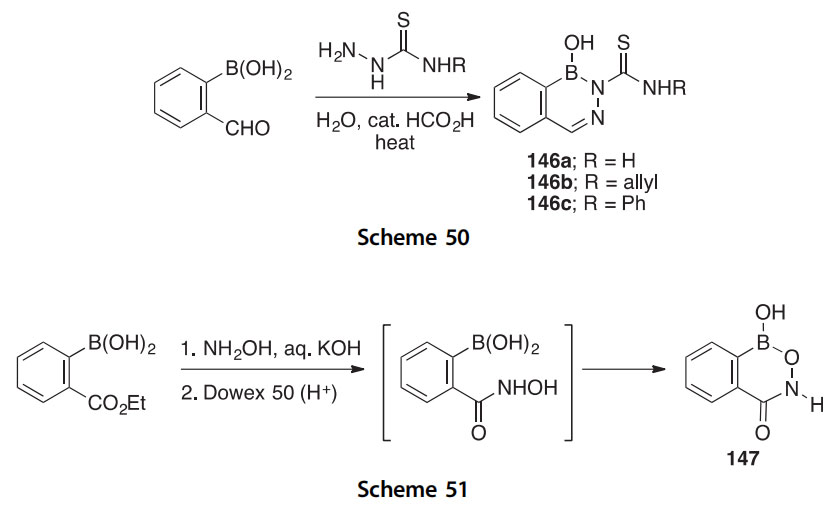
In another report on the condensation of phenylboronic acids, this time with thiosemicarbazides, heterocycles 146aec were synthesized and charac- terized by methods including crystallography (Scheme 50) (2008CB2415). Heterocycles 146aeb have shown appreciable activity against four fungi, but interestingly the derivative 146c did not.
The mode of intramolecular cyclization (5- vs 6-membered ring formation, O vs N attack) of a phenylboronic acid equipped with an ortho- hydroxamic acid group was determined to be a dehydration leading to the benzo[d][1,2,6]oxazaborinin-4(3H)-one 147 (Scheme 51) (2013AX(C)183). By X-ray, the boron heterocycle 147 is planar and displays both a phenol- like OH group and a lactam group. Strong intermolecular OeH/O]C and NeH/O]C interactions were noted in its crystal structure.
The set of 2-thiocarbamoylated 2,3,1-benzodiazaborines 148aeh, synthesized from 2-formylphenylboronic acids and thiosemicarbazides, was sought for potential antifungal activity (Scheme 52) (2015AJC366). Of the eight compounds prepared, four showed at least some activity against
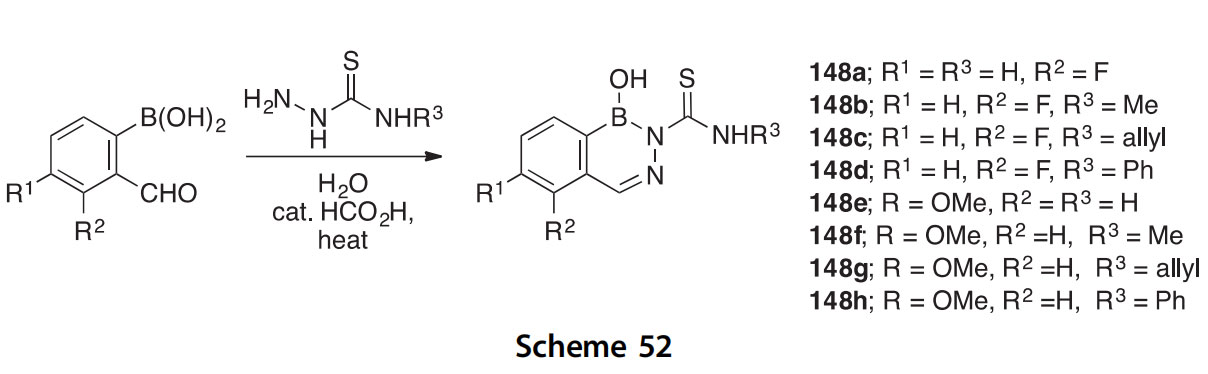
one or more of the five fungal species tested. X-ray crystal structures of products 148b, 148d, and 148h were obtained.
4. TRIHETERABOROLES AND TRIHETERABORINES
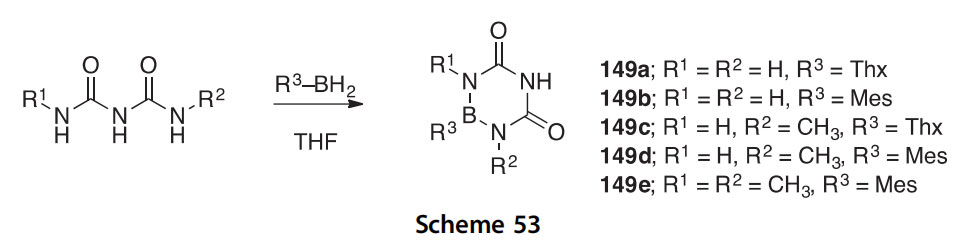
The boron heterocycles 149aee, which are 5-aza-6-bora analogs of uracil/thymine, were synthesized in a straightforward manner from substituted biurets and either thexylborane or mesitylborane (Scheme 53) (2010OL3386). Target 149a could not be isolated, 149b is stable, 149c was found to slowly decompose, and 149d,e are highly air- and moisture- stable. X-ray crystal structures of both 149b and 149d were determined.
5-, 6-, and 7-Membered cyclic b-enaminones have been shown to react with 4-substituted benzenediazonium tetraphenylborates to form the bicyclic [1,3,2l4]oxazaborines 150. These rearrange on heating to their 2H-[1,2,4,3l4]triazaborine isomers 151 which are fluorescent, especially in the solid state (emission lmax 541e592 nm) (Scheme 54) (2012JOM75). Compounds 150 and 151 have been characterized by multinuclear (1H, 13C, 11B, 15N) NMR, and X-ray crystal structures of representative compounds (150 and 151, both R X Me) have been determined. Compounds like 151 can be used for OLEDs. A similar set of oxazaborines and diazaborines were derived from b-enaminonitriles (2012T2052).
Trisubstituted oxadiazoboroles 152 were synthesized in 28e85% yields using the cyclocondensation reactions of thiophene, furan, or pyridine
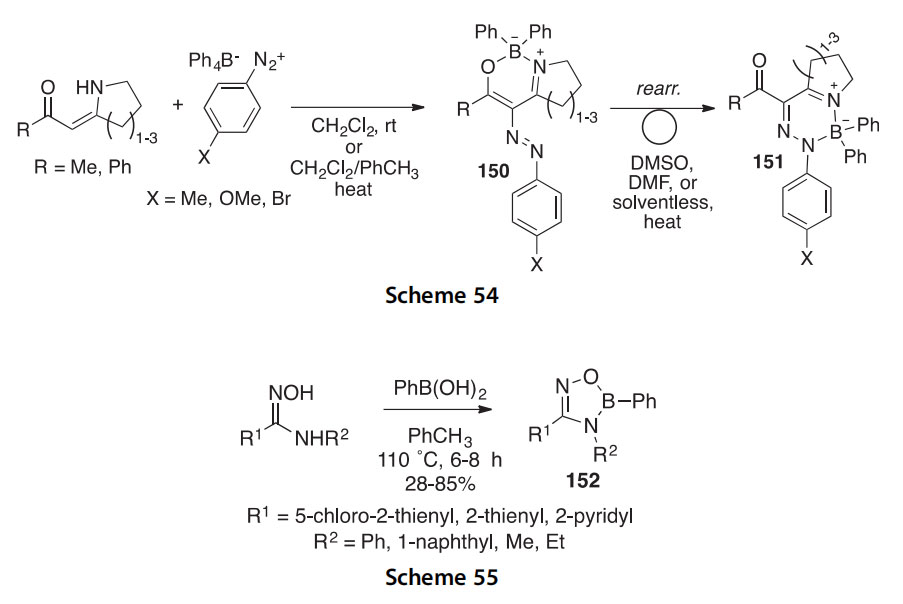
carboxamidoximes with phenylboronic acid in refluxing toluene (Scheme 55) (2008POL999).
Oxazaborines 153aeh have been synthesized by reaction of tetraphenyl- borates with b-enaminones and found to gradually rearrange to the triazabor- ines 154a,b at temperatures above 100 ○C (Scheme 56) (2006OM2025). The intermediates 155, suspected to be an equilibrium mixture of tautomers, were observed using 1H NMR and found to eliminate benzene before ring closure. In an extension of this study, the product mixture of the reaction between enaminoamides and tetraphenylborates depended upon the presence or absence of base (2009JOM63). In the absence of NaOAc, the product mix- tures of this reaction included oxazaborines (156aed), diazaborines (158a,b), and triazaborines (158aec). In the presence of NaOAc, the reaction mixtures consisted only of 156a, 156b, 158a, 159a, and 159b. The oxazaborine prod- ucts thermally rearrange to the diazaborine and triazaborine compounds in refluxing DMF.
5. TETRAHETERABOROLES AND TETRAHETERABORINES
Reaction of formazans with B(OAc)3 affords 2,3,5,6,1-tetrazaborines (boratatetrazines) 160a,b as dark purple solids in good yields (Scheme 57)

(2007CC126). X-ray crystal structures of the derivatives 160a,b were obtained, and using cyclic voltammetry it was shown that they can be reduced to borataverdazyl radical anions, the first-ever boron-containing verdazyl radicals.
6.BORAZINES AND BOROXINS
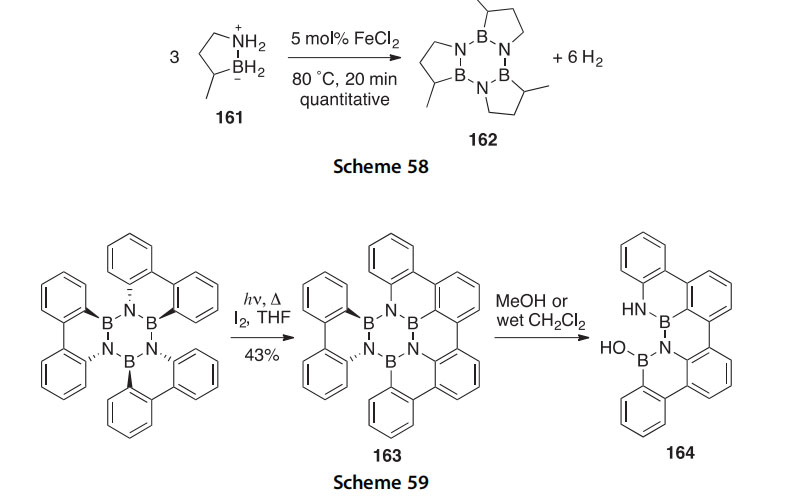
The synthesis and remarkable metal-catalyzed dehydrogenation of 3-methyl-1,2-azaborolidine 161 has been reported (Scheme 58) (2011JA19326). Compound 161 releases hydrogen quickly and efficiently, producing the borazine 162 without a phase change. This makes 161 attrac- tive as a storage material for hydrogen fuel. A method for converting bora- zine 162 back to 161 in 92% overall yield using LiAlH4 as a reductant has also been described.
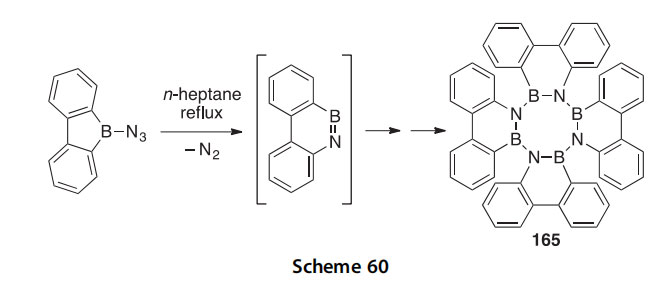
An approach that has not received as much attention as conventional synthetic methods, but which may prove useful in the synthesis of BN PAH analogs, is the method of photocyclization. The phenanthrene annelated B3N3 tribenzoperylene 163 was synthesized in 43% yield after 1,2:3,4:5,6-tris(o,o0-biphenylylene)borazine was irradiated with light of 280e400 nm in the presence of iodine (Scheme 59) (2014CC7821). The reaction is assumed to proceed via the formation of an intermediate trans- dihydro derivative that is then oxidized by I2 to form 163. When treated with MeOH or wet CH2Cl2, 163 is hydrolyzed to 164.
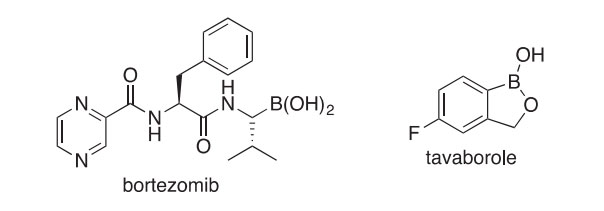
The tetramer (165) of a B-N-phenanthryne was isolated from a complex mixture of products formed by the thermolysis of 9-azido-9-borafluorene